Example: alignment of calmodulin
Calmodulin is a calcium binding protein. It is composed of two similar domains, each of which binds two calcium atoms. The protein is especially flexible, which can make structure alignment challenging. Here we will compare the calcium binding loops of two calmodulin crystal structures - PDB codes 1A29 and 4G28.Initial global alignment The 1A29 project is opened and the Align Protein Structure dialog is filled out as in figure 12.21. Selecting "All chains from 1A29" tells the aligner to make the best possible global alignment, favoring no particular region. The output of the alignment is shown in figure 12.22. The blue chain is from 1A29, the brown chain is the corresponding calmodulin chain from 4G28 (a calmodulin-binding chain from the 4G28 file has been hidden from the view). Because calmodulin is so flexible, it is not possible to align both of its domains (enclosed in black boxes) at the same time. A good global alignment would require the brown protein to be translated in one direction to match the N-terminal domain, and in the other direction to match the C-terminal domain (see black arrows).
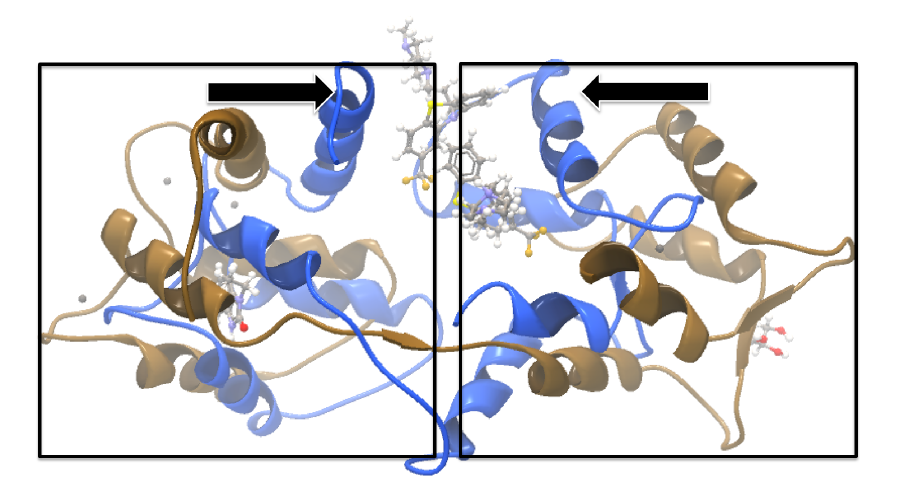
Figure 12.22: Global alignment of two calmodulin structures (blue and brown). The two domains of calmodulin (shown within black boxes) can undergo large changes in relative orientation. In this case, the different orientation of the domains in the blue and brown structures makes a good global alignment impossible: the movement required to align the brown structure onto the blue is shown by arrows - as the arrows point in opposite directions, improving the alignment of one domain comes at the cost of worsening the alignment of the other.
Focusing the alignment on the N-terminal domain To align only the N-terminal domain, we return to the 1A29 project and select the Show Sequence action from beneath the Project Tree. We highlight the first 62 residues, then convert them into an atom group by right-clicking on the "Current" selection in the Project Tree and choosing "Create Group from Selection" (figure 12.23). Using the new atom group as the reference in the alignment dialog leads to the alignment shown in figure 12.24. In addition to the original input proteins, the output now includes two Atom Groups, which contain the atoms on which the alignment was focused. The History of the output Molecule Project shows that the alignment has 0.9 Å RMSD over the 62 residues.
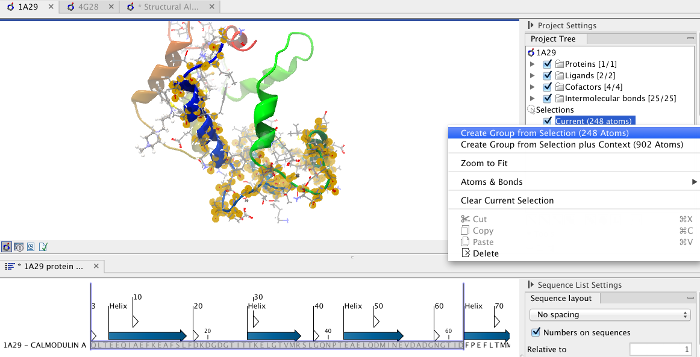
Figure 12.23: Creation of an atom group containing the N-terminal domain of calmodulin.
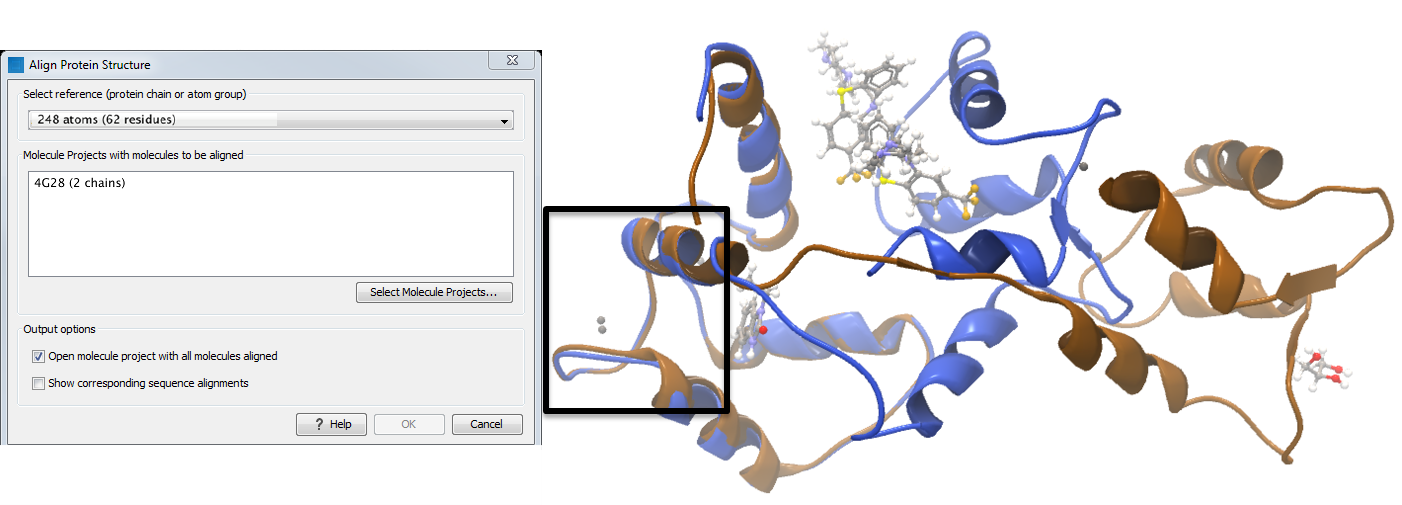
Figure 12.24: Alignment of the same two calmodulin proteins as in figure 12.22, but this time with a focus on the N-terminal domain. The blue and brown structures are now well-superimposed in the N-terminal region. The black box encloses two calcium atoms that are bound to the structures.
Aligning a binding site Two bound calcium atoms, one from each calmodulin structure, are shown in the black box of figure 12.24. We now wish to make an alignment that is as good as possible about these atoms so as to compare the binding modes. We return to the 1A29 project, right-click the calcium atom from the cofactors list in the Project Tree and select "Create Nearby Atoms Group". Using the new atom group as the reference in the alignment dialog leads to the alignment shown in figure 12.25.
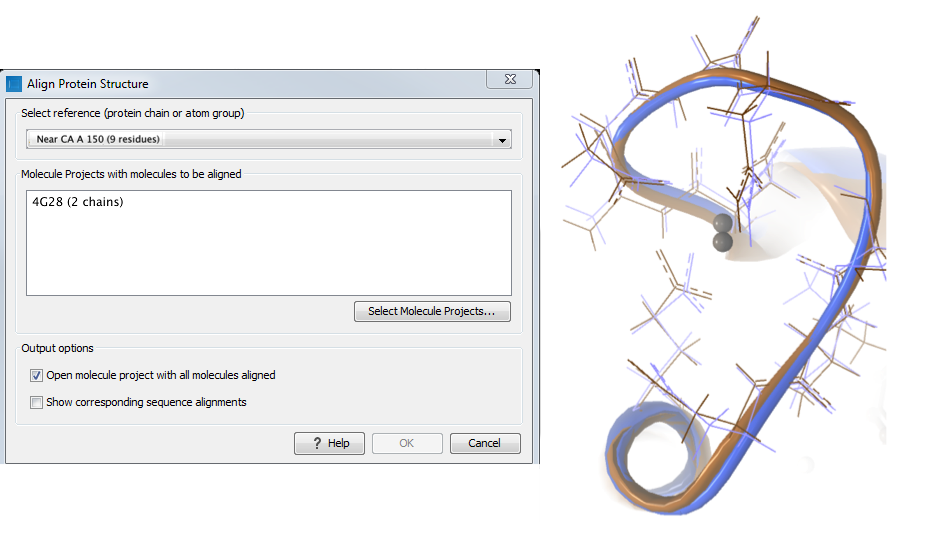
Figure 12.25: Alignment of the same two calmodulin domains as in figure 12.22, but this time with a focus on the calcium atom within the black box of figure 12.24. The calcium atoms are less than 1 Å apart - compatible with thermal motion encoded in the atoms' temperature factors.