Trim Primers and their Dimers from Mapping
Trim Primers and their Dimers from Mapping unaligns the primer parts of reads in read mappings, and also unaligns parts of reads identified as primer dimerization artifacts. Unaligned regions are not considered by downstream tools such as variant callers, where it would be undesirable to consider primer regions.
Trim Primers and their Dimers from Mapping was designed for use with data generated using GeneReadTM DNAseq Targeted Panels V2, where target specific primer pairs are used for multiplexed PCR-based target enrichment. We expect the tool to work with other targeted amplicon sequence data that employ target specific primer pairs, but we have not tested it for that purpose. This tool is included in the QIAGEN GeneRead Panel Analysis template workflow, where the relevant workflow element has been named "Trim Primers and their Dimers of Mapped Reads".
Primer trimming
Target primer locations need to be imported before using this tool. Importing descriptions of primer locations from a generic text format file or from a QIAGEN gene panel primer file is described in the Import Primer Pairs section of the CLC Genomics Workbench manual: http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Import_Primer_Pairs.html.
The fraction of the primer that must overlap with a read's aligned bases in order to record a primer hit is configurable.
By default, reads are only retained in the mapping if primer sequence is detected at the start of the read, but this behavior is optional.
For paired end data, if the primer found on R2 is not a member of the same target region primer pair as the primer found on R1, both members of the pair will be removed from the mapping.
Trim Primers and their Dimers from Mapping is strict regarding primer position. Primers are expected at the 5' end or the 3' end of the read, whether the end is aligned or unaligned. If there are any additional bases at the 5' or 3' end, the region will not be identified as primer sequence.
Primer dimer trimming
Two steps are involved in trimming primer dimerization artifacts:
- All primers are compared against all others to look for pairs likely to dimerize. The minimum number of overlapping bases, used to identify primers that may dimerize with each other, is configurable. A list is compiled containing, for each primer, all primers with potential to dimerize with it.
- If a primer p has been trimmed (unaligned), and the still-mapped section of the affected read starts with the sequence of a primer identified as dimerizing with p, it is assumed that the read contains primer-dimer artifact. This predicted dimerization artifact is then unaligned. If a read only contains primer-dimer artifact, the read is removed from the mapping and discarded.
The Trim Primers and their Dimers from Mappings tool also includes an option for trimming primers of amplicon fragments. This is particularly useful for trimming reads originating from short fragments. The Trim primers of amplicon fragments option allows trimming of both forward and reverse primers of reads in both directions. In cases where primers overlap, the innermost primer is used for trimming irrespective of the read orientation and expected primer pairing. In other words, the primer ID is ignored when selecting the Trim primers of amplicon fragments option. This allows trimming of reads that end in a region with multiple overlapping primers, where it cannot be determined which primer the read originated from and consequently how much of the read end is primer sequence.
Running the tool
To launch Trim Primers and their Dimers from Mapping, go to:
Toolbox | Resequencing Analysis (![]() ) | Trim Primers and their Dimers from Mapping (
) | Trim Primers and their Dimers from Mapping (![]() )
)
In the first wizard step (figure 19.12), you are asked to select a read mapping. If you would like to analyze more than one read mapping, you can run the analysis in batch mode by ticking the "Batch" box in the lower left corner of the wizard. Running jobs in batch mode is described in the CLC Genomics Workbench manual: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Standard_batch_processing.html.
Figure 19.12: Select a read mapping.
In the next wizard step (figure 19.13), settings for the tool are configured.
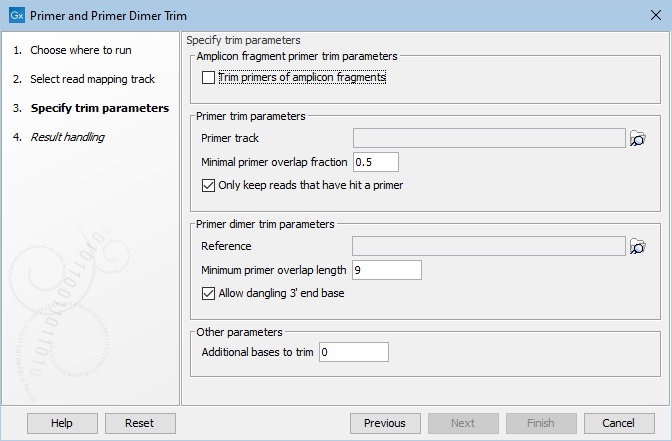
Figure 19.13: Select your primer location file and choose whether you want to keep or discard reads with no matching primers. The option Trim primers of amplicon fragments is useful when working with reads that originate from short fragments.
- Amplicon fragment primer trim parameters
- Trim primers of amplicon fragments If you tick "Trim primers of amplicon fragments" all reads, regardless of orientation, can be trimmed with both forward and reverse primers.
- For read pairs mapping in the forward orientation (dark blue color) trim reads if:
- 5' end of Read 1 starts within a forward primer annotation
- 5' end of Read 2 starts within a reverse primer annotation
- 3' end of Read 2 ends within a forward primer annotation
- 3' end of Read 1 ends within a reverse primer annotation
- For read pairs mapping in the reverse orientation (light blue color) trim reads if:
- 5' end of Read 2 starts within a forward primer annotation
- 5' end of Read 1 starts within a reverse primer annotation
- 3' end of Read 1 ends within a forward primer annotation
- 3' end of Read 2 ends within a reverse primer annotation
- For read pairs mapping in the forward orientation (dark blue color) trim reads if:
- Primer trim parameters
- Primer track Click on the folder icon on the right-hand side of the wizard to select your primer location file.
- Minimal primer overlap fraction Specifies the fraction of the primer that must overlap with the read's aligned bases in order to record a primer hit. Setting the fraction to 0.0 will disable this requirement.
- Read handling configuration If you tick "Only keep reads that have hit a primer", reads with no matching primers will be discarded.
- Primer dimer trim parameters
- Reference Click on the folder icon on the right-hand side of the wizard to select your reference location file.
- Minimum primer overlap length The minimum number of bases that need to bind for primers to dimerize and amplify.
- Allow dangling 3' end base If you tick "Allow dangling 3' end base", a mismatch is allowed in the primer dimerization at the 3' end.
- Other parameters
- Additional bases to trim This number of nucleotides will be trimmed off a read right after the primer. This trimming is not done on reads for which primer-dimer artifacts were identified. This is set by default to 2 to avoid false positive calls and increase accuracy of the coverage calculation in the report.
- Trim primers of amplicon fragments If you tick "Trim primers of amplicon fragments" all reads, regardless of orientation, can be trimmed with both forward and reverse primers.
In the last wizard step, choose the results to save and and click on Finish.
Output of Trim Primers and their Dimers from Mapping
The default output is a read mapping with primer and primer-dimer regions of the mapped reads unaligned. The name of the read mapping generated is based on the name of the mapping used as input, with "trimmed reads" appended. Optionally, a track containing the primer-dimer regions used for trimming the reads will also be generated. This track contains information about why each primer-dimer pair was predicted and the number of times it was used to partially trim a read or to remove a read from the mapping because it consisted only of primer-dimer sequence.