Perform QIAseq Immune Repertoire Analysis template workflow
Using RNA-Seq data as input, the Perform QIAseq Immune Repertoire Analysis workflow can be used to characterize the T cell receptor (TCR) repertoire.
The workflow includes all necessary steps for processing the RNA-Seq reads and characterizing the repertoire:
- UMIs are removed using Remove and Annotate with Unique Molecular Index;
- UMI reads are created using Create UMI Reads from Reads;
- Overlapping paired reads are merged using Merge Overlapping Pairs;
- Reads are trimmed using Trim Reads;
- Clonotypes are identified using Immune Repertoire Analysis;
- Reports are combined using Create Sample Report.
Note that the workflow has been designed and configured for data produced with the QIAseq Immune Repertoire RNA Library Kit. It can, however, be used as a starting point to analyze other types of data, for example to characterize the B cell receptor (BCR) repertoire. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Editing_existing_workflows.html.
To run Perform QIAseq Immune Repertoire Analysis, go to the toolbox and select:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | Other QIAseq Workflows (
) | Other QIAseq Workflows (![]() ) | Perform QIAseq Immune Repertoire Analysis (
) | Perform QIAseq Immune Repertoire Analysis (![]() )
)
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
Select the RNA-Seq reads in the input dialog. When analyzing more than one sample at the time, the Batch checkbox in the lower left corner of the dialog should be checked.
In the next step you need to specify the relevant Reference Data Set. The Reference Data Manager (see Reference Data Management) offers two QIAGEN sets:
- QIAseq Immune Repertoire Analysis for analysis of TCR human data.
- QIAseq Immune Repertoire Analysis Mouse for analysis of TCR mouse data.
The reference data should be downloaded to the navigation area prior to analysis. If you have not downloaded the Reference Data Set, the dialog will suggest the relevant data set(s) and offer the opportunity to download it (see figure 8.3).
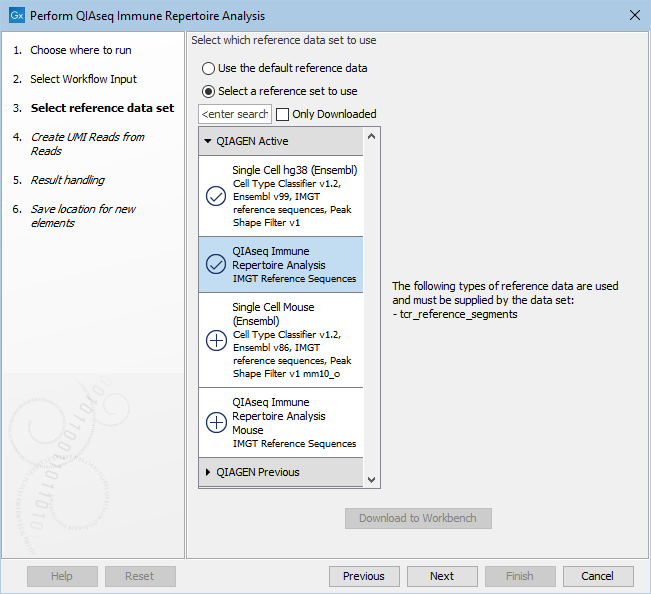
Figure 8.3: The human Reference Data Set is highlighted. The references needed by the workflow are listed to the right, and the relevant reference data sets are listed to the left.
In the example shown in figure 8.3, the check mark to the left of the human reference data indicates that it has already been downloaded and is ready to use. The plus symbol next to the mouse reference data indicates that it has not been downloaded. To download the mouse data, click on the QIAseq Immune Repertoire Analysis Mouse text to enable the download button. The option "Use the default reference data" is available when the corresponding Reference Data Set, here human, is already downloaded. You can always use the "Select a reference data set to use" to specify another Reference Data Set. You can inspect which is the default reference data in the "Use the default reference data" tool-tip.
When running the workflow in Batch mode, a dialog will now appear where the batch units should be defined. If metadata is not needed, choose 'Use organization of input data', otherwise choose 'Use metadata' and select a metadata table. Information about metadata creation / import and usage can be found here http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Metadata.html. The next dialog will show an overview of the defined batch units.
In the next dialog the 'Minimum UMI group size' can be set, which specifies the minimum size of a UMI group. Reads belonging to UMI groups supported by fewer reads than this number will be discarded. See Remove and Annotate with Unique Molecular Index and Create UMI Reads from Reads for information about UMI identification and processing.
Finally, choose where to save the data, and press Finish to start the analysis.
Subsections