- Run the Identify and Annotate Variants (TAS) workflow
- Output from the Identify and Annotate Variants (TAS) workflow
Identify and Annotate Variants (TAS)
The Identify and Annotate Variants (TAS) template workflow should be used to identify and annotate variants in one sample. The workflow is a combination of the Identify Variants and the Annotate Variants workflows.
The workflow starts with mapping the sequencing reads to the human reference sequence, followed by a local realignment to improve the variant detection that is run afterwards. After the variants have been detected, they are annotated with gene names, amino acid changes, conservation scores, information from relevant variants present in the ClinVar database, and information from common variants present in the common dbSNP Common, HapMap, and 1000 Genomes database. Furthermore, a detailed targeted region coverage report is created to inspect the overall coverage and mapping specificity.
Before starting the workflow, you will need to import in the workbench a file with the genomic regions targeted by the amplicon or hybridization kit. Such a file (a BED or GFF file) is usually available from the vendor of the enrichment kit and sequencing machine. Use the Import | Tracks tool to import it in your Navigation Area.
Run the Identify and Annotate Variants (TAS) workflow
To run the Identify and Annotate Variants (TAS) workflow, go to:
Toolbox | Template Workflows | Biomedical Workflows (![]() ) | Targeted Amplicon Sequencing (
) | Targeted Amplicon Sequencing (![]() ) | Somatic Cancer (
) | Somatic Cancer (![]() ) | Identify and annotate Variants (TAS) (
) | Identify and annotate Variants (TAS) (![]() )
)
- Double-click on the workflow name to start the analysis. If you are connected to a server, you will first be asked where you would like to run the analysis.
- First select the sequencing reads from the sample that should be analyzed (figure 24.34).
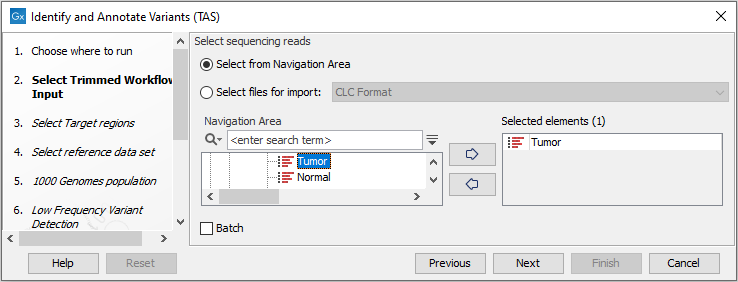
Figure 24.34: Select all sequencing reads from the sample to be analyzed.If several samples should be analyzed, the tool has to be run in batch mode. This is done by checking "Batch" and selecting the folder that holds the data you wish to analyze.
- In the Target regions dialog (figure 24.35), you
can specify the target regions track. Variants will only be called in the target regions.
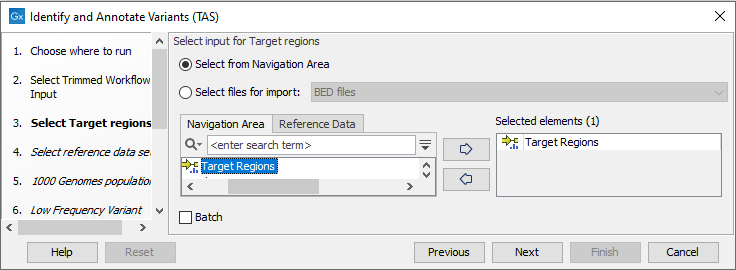
Figure 24.35: In this wizard step you can specify the target regions track. Variants will not be called outside these regions. - In the next dialog, you have to select which reference data set should be used in the analysis (figure 24.36).
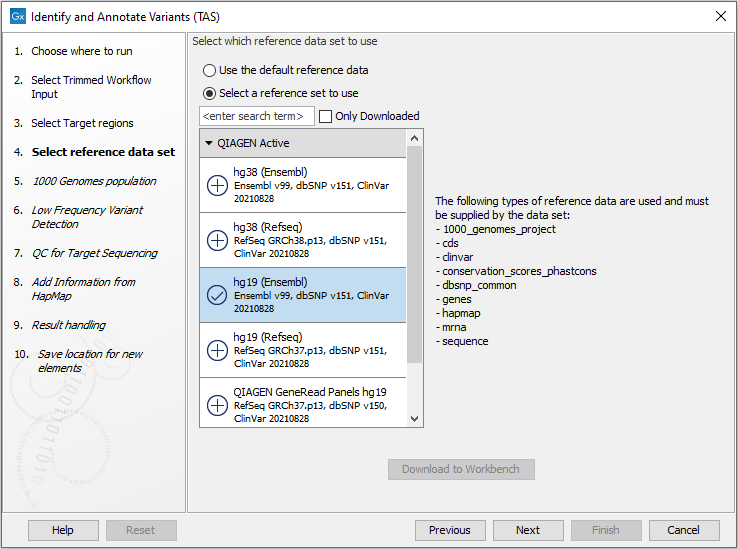
Figure 24.36: Choose the relevant reference Data Set to identify and annotate. - In the next wizard step (figure 24.37) you can select the population from the 1000 Genomes project that you would like to use for annotation.
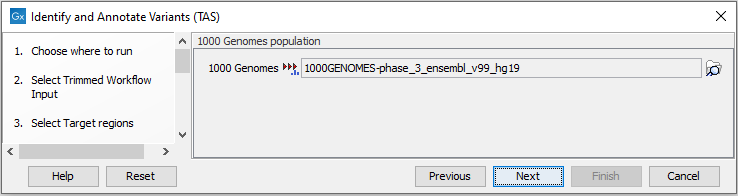
Figure 24.37: Select the population from the 1000 Genomes project that you would like to use for annotation. - In the next dialog (figure 24.38), you have to specify the parameters for the variant detection.
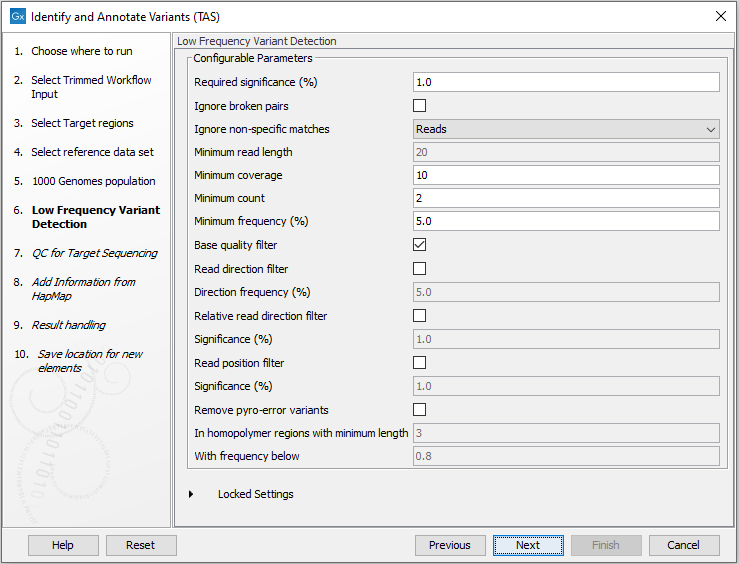
Figure 24.38: Specify the parameters for variant calling.For a description of the different parameters that can be adjusted, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Low_Frequency_Variant_Detection.html. If you click on "Locked Settings", you will be able to see all parameters used for variant detection in the template workflow.
- In the QC for Target Sequencing step (figure 24.39) you can specify the minimum read coverage that should be present in the targeted regions.
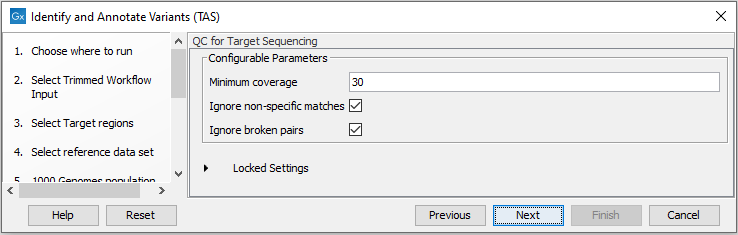
Figure 24.39: Set the parameters for the QC for targeted regions. - Finally, select a population from the HapMap database (figure 24.40). This will add information from the Hapmap database to your variants.
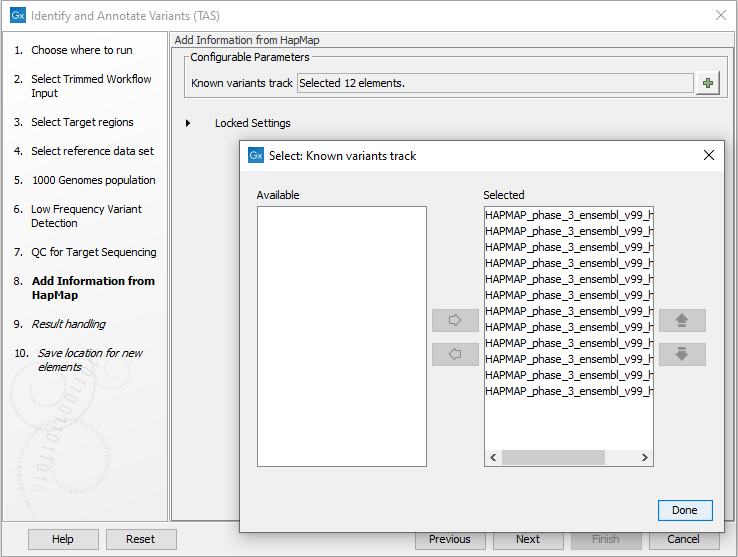
Figure 24.40: Select a population from the HapMap database to add information from the Hapmap database to your variants. - In the last wizard step you can check the selected settings by clicking on the button labeled Preview All Parameters.
In the Preview All Parameters wizard you can only check the settings, and if you wish to make changes you have to use the Previous button from the wizard to edit parameters in the relevant windows.
- Choose to Save your results and click Finish.
Output from the Identify and Annotate Variants (TAS) workflow
The Identify and Annotate Variants (TAS) tool produces several outputs.
Please do not delete any of the produced files alone as some of them are linked to other outputs. Please always delete all of them at the same time.
A good place to start is to take a look at the mapping report to see whether the coverage is sufficient in the regions of interest (e.g. > 30 ). Furthermore, please check that at least 90% of the reads are mapped to the human reference sequence. In case of a targeted experiment, please also check that the majority of the reads are mapping to the targeted region.
Next, open the Track List file (see figure 24.41).
The Track List includes a track of the identified annotated variants in context to the human reference sequence, genes, transcripts, coding regions, targeted regions, mapped sequencing reads, relevant variants in the ClinVar database as well as common variants in common dbSNP Common, HapMap, and 1000 Genomes databases.
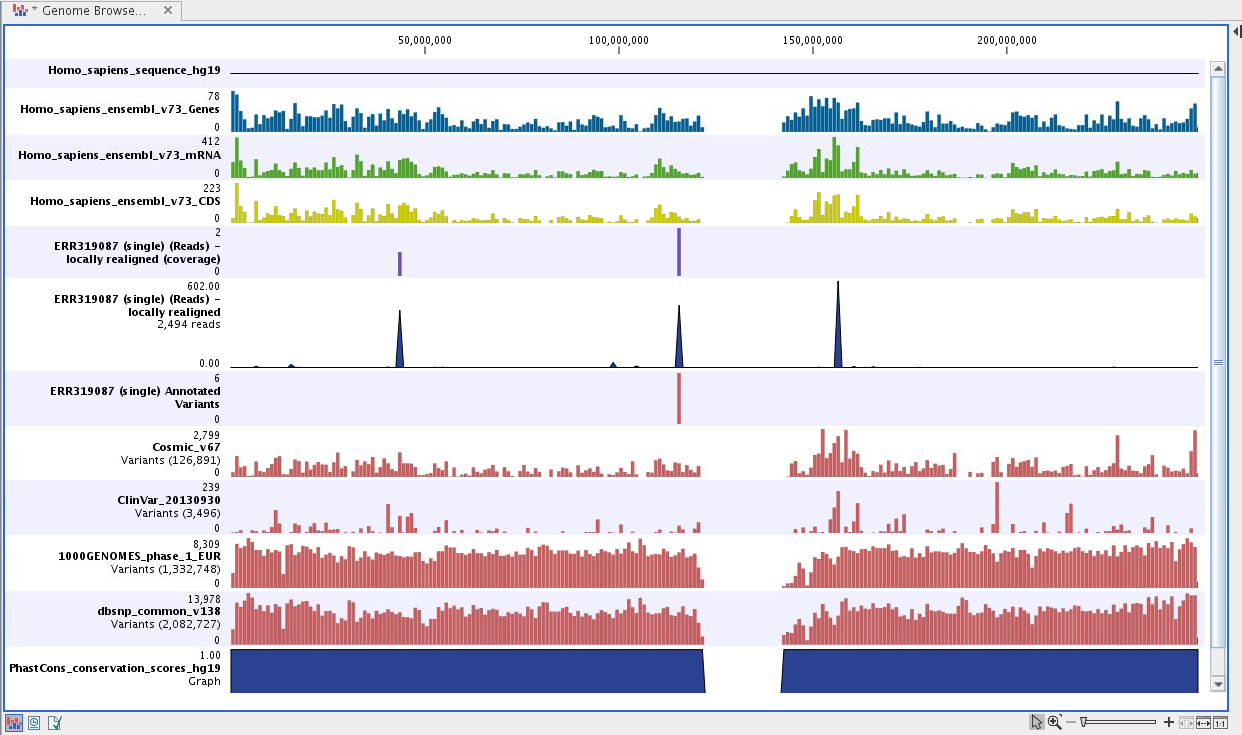
Figure 24.41: Track List to inspect identified variants in
the context of the human genome and external databases.
To see the level of nucleotide conservation (from a multiple alignment with many vertebrates) in the region around each variant, a track with conservation scores is added as well.
By double-clicking on the annotated variant track in the Track List, a table will be shown that includes all variants and the added information/annotations (see figure 24.42).
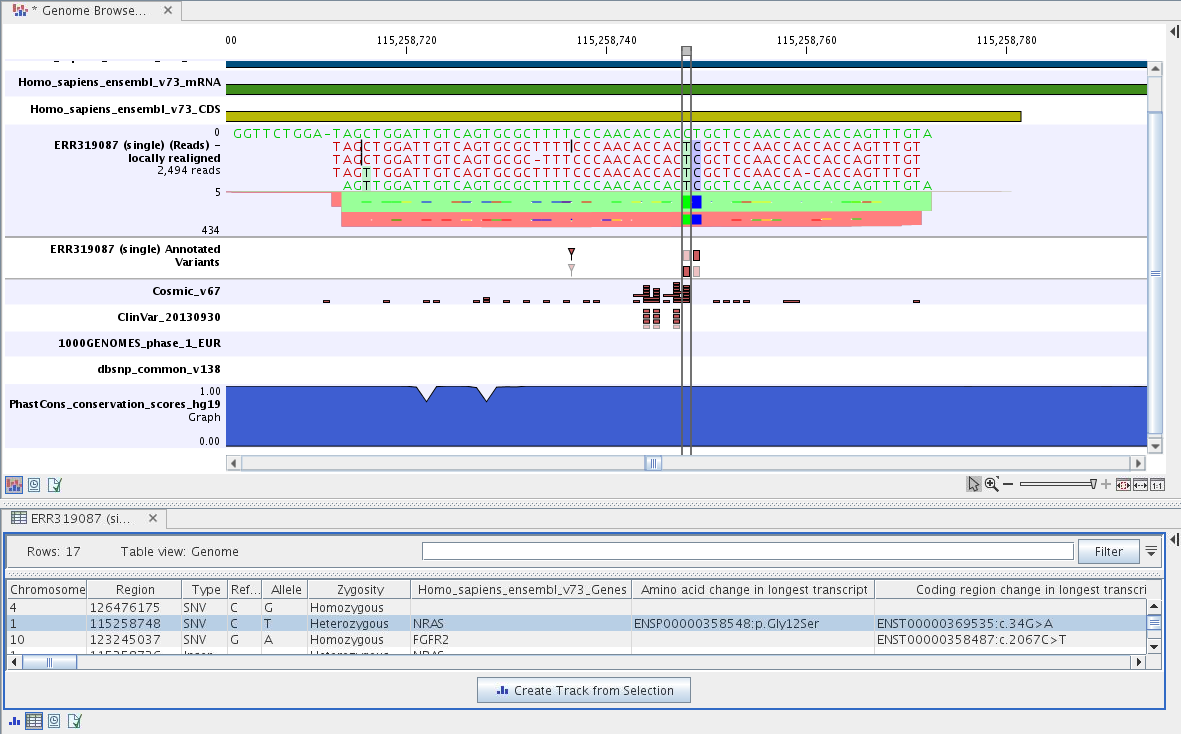
Figure 24.42: Track List with an open track table to
inspect identified somatic variants more closely in
the context of the human genome and external databases.
The added information will help you to identify candidate variants for further research. For example can common genetic variants (present in the HapMap database) or variants known to play a role in drug response or other relevant phenotypes (present in the ClinVar database) easily be seen.
Not identified variants in ClinVar, can for example be prioritized based on amino acid changes (do they cause any changes on the amino acid level?). A high conservation level on the position of the variant between many vertebrates or mammals can also be a hint that this region could have an important functional role and variants with a conservation score of more than 0.9 (PhastCons score) should be prioritized higher. A further filtering of the variants based on their annotations can be facilitated using the table filter on top of the table.
If you wish to always apply the same filter criteria, the "Create new Filter Criteria" tool should be used to specify this filter and the "Identify and Annotate" workflow should be extended by the "Identify Candidate Tool" (configured with the Filter Criterion). See the reference manual for more information on how preinstalled workflows can be edited.
Please note that in case none of the variants are present in ClinVar or dbSNP Common, the corresponding annotation column headers are missing from the result.
In case you like to change the databases as well as the used database version, please use the Reference Data Manager.