Import your reads
Use the appropriate Import button to import the reads in the Navigation Area of the Workbench.
Click on the folder icon (![]() ) to the right of the "Select files" field, and select the files you want to import.
) to the right of the "Select files" field, and select the files you want to import.
The Illumina High-Throughput Sequencing Import offers the following options (figure 5.6):
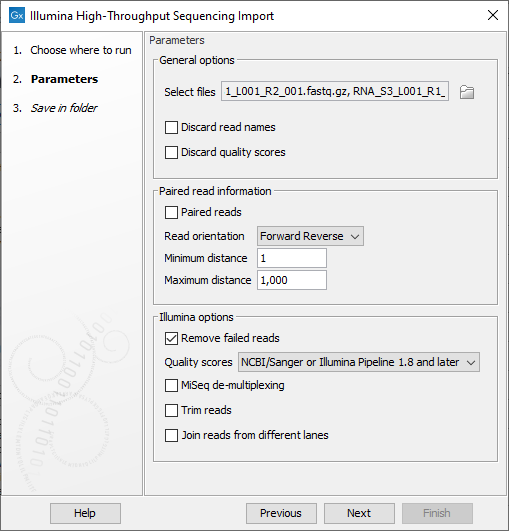
Figure 5.6: The Analyze QIAseq Samples Illumina Import dialog.
- General options
- Discard sequence names. For high-throughput sequencing data, the naming of the individual reads is often irrelevant given the amount of reads. This option allows you to discard read names to save disk space.
- Discard quality scores. Quality scores are visualized in the mapping view and they are used for variant and fusion detection. Therefore, we do not recommend to discard the quality scores during import.
- Paired read information
- Paired-end reads. Check the option if the reads are paired, then choose their orientation (Forward Reverse for end-paired or Reverse Forward for mate-pair) in the drop down menu.
- Set minimum and maximum distances. The paired read distance includes the full read sequence, which means that is from the beginning of the forward read to the beginning of the reverse read. The distances are usually defined during the library preparation of your sequencing experiment, but in doubt you can enter default values: for paired-end reads, distances are between 1 and 1000 bp while mate-pair reads typically have longer distances between 1000-5000 bp (and sometimes up to 10000).
- Illumina options
- Remove failed reads. If you check Remove failed reads, reads that did not pass a quality filter (in qseq and fastq files) will be ignored during import. For more information on format specific quality filters see section on file format above). If you import paired data and one read in a pair is removed during import, the remaining mate will be saved in a separate sequence list with single reads.
- Quality score. Choose which Illumina pipeline version was used to sequence your reads.
- MiSeq de-multiplexing. Using this option on MiSeq multiplexed data will divide reads into different files based on the "IndexSequence" of the read header:
@Instrument:RunID:FlowCellID:Lane:Tile:X:Y:UMI ReadNum:FilterFlag:0:IndexSequenceSubsequent analysis can then be executed in batch on all the files, and results can be compared at the end.
- Illumina trim. This option applies to Illumina Pipeline 1.5 to 1.7. In this pipelines, the Phred scale (ASCII 64 to 104) disregards value 0 and 1, but the value 2 (B) has special meaning and is used as a trim clipping. This means that when selecting Illumina Pipeline 1.5 and later, the reads are trimmed when a B is encountered at either end of the reads in the input file if the Trim reads option is checked.
- Join reads from different lanes. Fastq files from the same sequencing run but from different lanes will be merged into a single sequences list.
- Remove failed reads. If you check Remove failed reads, reads that did not pass a quality filter (in qseq and fastq files) will be ignored during import. For more information on format specific quality filters see section on file format above). If you import paired data and one read in a pair is removed during import, the remaining mate will be saved in a separate sequence list with single reads.
The Ion Torrent High-Throughput Sequencing Import (figure 5.7) just requires you to specify a SAM or BAM file containing unmapped reads.
Figure 5.7: The Analyze QIAseq Samples Ion Torrent Import dialog.
Note that if your Ion Torrent reads are already mapped, they will be ignored by this importer, and a warning dialog will state that the SAM/BAM file contained more references than selected references, followed by a list of missing chromosomes and the number of reads aligning to these chromosomes. To import correctly mapped Ion Torrent reads, exit the Analyze QIAseq Samples guide and use the Import | Ion Torrent button in the Toolbar of the Workbench.