Output from the Structural Variant Caller
The tool produces the following outputs:- Indels (Indels) A variant track with indels (deletions and insertions (including tandem duplications)) that have lengths up to 100,000 bp
- Long indels (Indels long)) An annotation track with long indels (those with lengths larger than 100,000 bp)
- Inversions (Inv) An annotation track with inversions
- Breakpoints (BP) An annotation track with a row for each breakpoint showing the unaligned ends used for the analysis
- Report A report giving an overview over analyzed references and found structural variants
Indels variant track
The indels track contains all the standard variant annotations, except for the "Probability" and "QUAL" columns which are only preduced when the Whole genome Sequencing application is chosen. When produced, the content of the "Probability" column is the average of the probabilities of the breakpoints used to infer the feature, and the content of the "QUAL" column is the Phred score version of that probability.As the indels are inferred indirectly from the unaligned ends, and hence are not necessarily directly visible within the aligned parts of the reads, the indel variant annotations are approximated from the breakpoints and the unaligned ends of reads in the read mapping. Figure 26.8 shows a read mapping of a 52bp deletion and the read mapping in which it was inferred by examination of the indirect evidence in the reads with unaligned ends, along with the approximated variant annotations (e.g count, coverage and frequency).
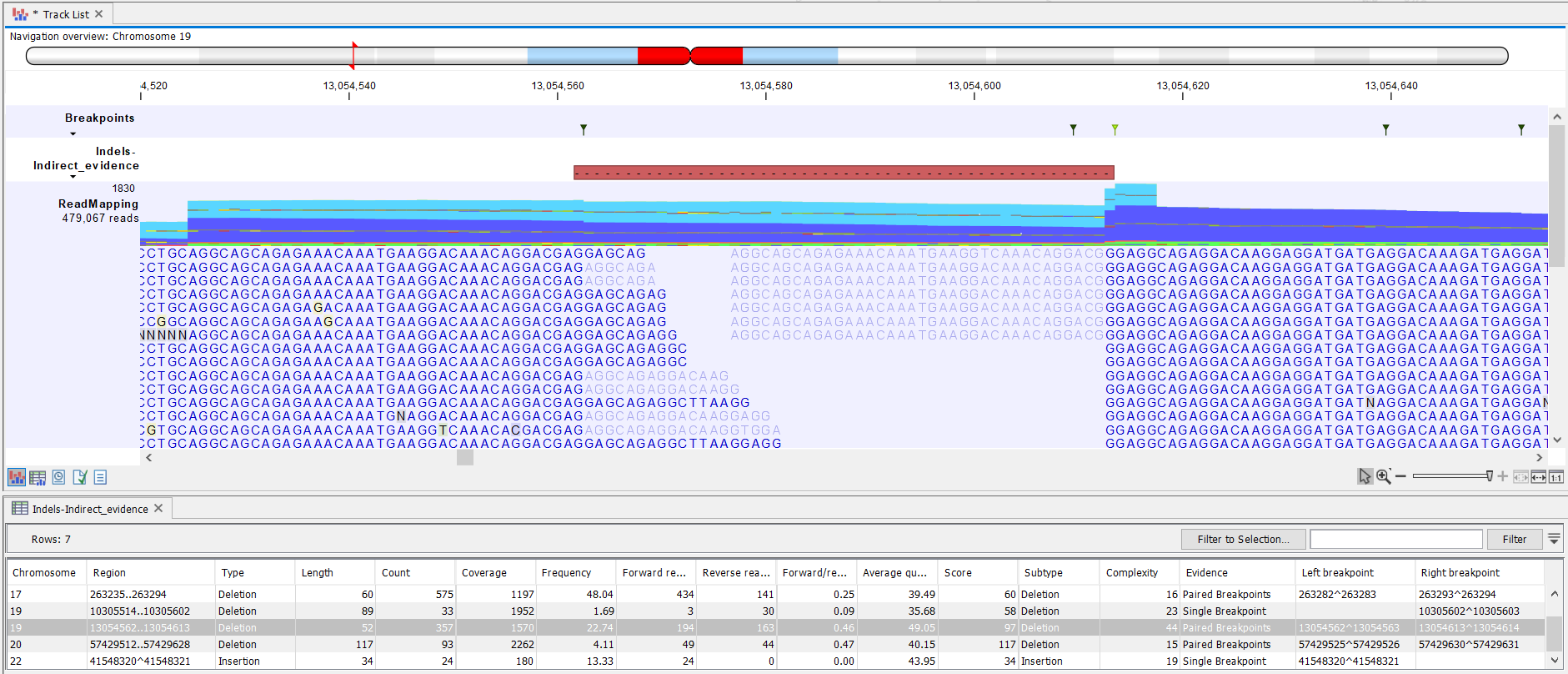
Figure 26.8: A 52 bp deletion with approximated variant annotations and the read mapping in which it was inferred.
In addition to the standard variant annotations, the indel track contains the following columns with characteristics of the inferred structural variant (figure 26.9):
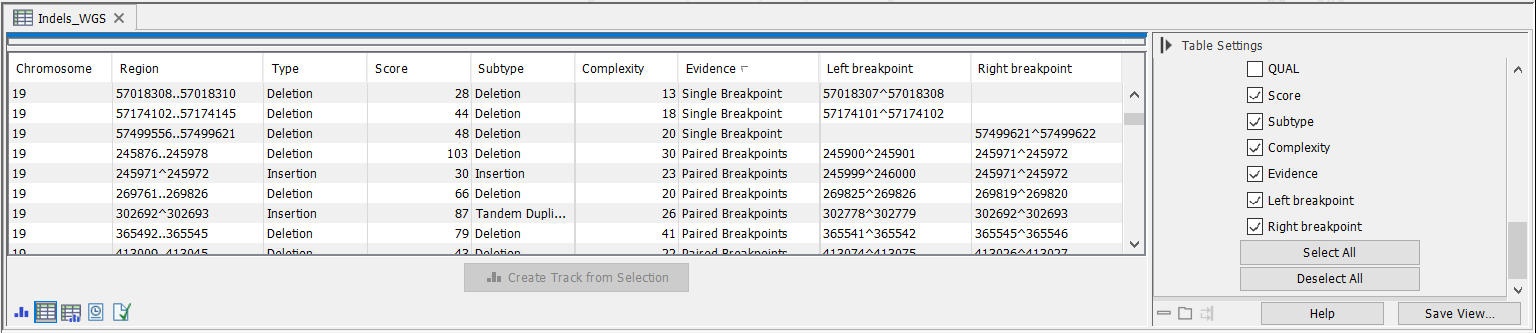
Figure 26.9: The Structural Variant Caller indels track.
- Score Measure of the overall evidence supporting the structural variant detected. The value is based on the alignment scores of the unaligned ends or, in case of shorter indels, the length of the variation.
- Subtype This is a more specific categorization of the structural variant type: either Insertion, Deletion, Tandem Duplication, Inversion, CNV Loss, or CNV Gain. Note that for Tandem Duplications only one duplication is reported, even in cases where a sequence appears in more than two copies in the reads. The CNV loss and CNV gain annotations are CNVs inferred from a combination of coverage and breakpoint evidence, and are only inferred for Whole genome Sequencing applications.
- Evidence May be either Single Breakpoint, Paired Breakpoints, CNV + Breakpoint (i.e., based on coverage information and a single breakpoint), or Broken pairs. The broken pairs option is special since it is based on assembly of broken read pairs, where one of the reads in a pair maps at a different location in the genome. This allows for detection of insertions of Alu elements for example.
- Complexity Sum of the complexity of the left and right unaligned ends.
- Left breakpoint Position of the left breakpoint of the structural variant.
- Right breakpoint Position of the right breakpoint of the structural variant.
Long indels and inversions annotation tracks
The long indels and inversions feature tracks contain the same columns as the indels track, except that the "Type", "Reference", "Allele" and "Reference allele" columns in the indels track are replaced by a single "Name" column in the feature track. The "Name" column specifies whether the feature is a deletion, insertion or inversion.
The report
The report (figure 26.10) gives an overview of the numbers and types of structural variants found in the sample.
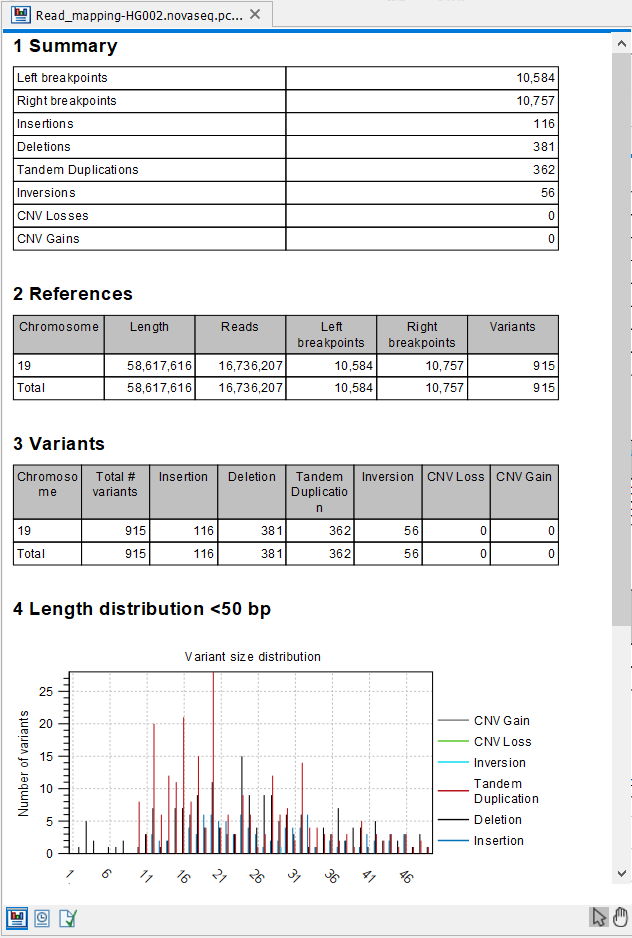
Figure 26.10: The Structural Variant Caller report.
It contains:
- A 'Summary' table giving an overview of the numbers of breakpoints identified, and numbers of the different types and subtypes of the structural variants found
- A 'References' table with a row for each reference sequence, and information on the number of left and right unaligned breakpoint signatures and the resulting number of structural variants found on that reference sequence.
- A 'Variants' table with a row for each reference sequence, and information on the total number of variants, stratified into the different variant categories (Insertion, Deletion, Tandem Duplication, Inversion, CNV Loss, CNV Gain) found on that reference sequence.
- A length distribution plot for short (<50 bp) structural variants
- A length distribution plot for long (>50 bp) structural variants
The report generated by this tool can be used with the Combine Reports tool (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Combine_Reports.html).
Breakpoint track (BP)
The breakpoint track (figure 26.11) contains a row for each called breakpoint with the following information
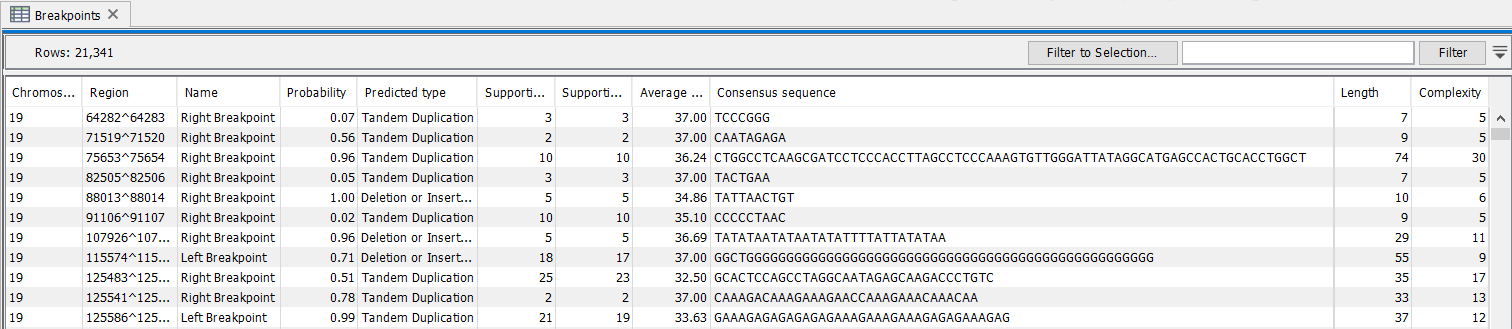
Figure 26.11: The Structural Variant Caller breakpoints track.
- Chromosome Chromosome on which the breakpoint is located.
- Region Location on the chromosome of the breakpoint.
- Name Type of the breakpoint ('left breakpoint' or 'right breakpoint').
- Probability Estimate for how trustworthy the prediction is when running on WGS data
- Predicted type Whether the breakpoint appears to be part of a tandem duplication, a deletion or insertion. This is an initial estimate that does not include the possibility of inversions and is used with WGS data.
- Supporting reads Number of reads at the breakpoint position with an unaligned end.
- Supporting reads (weighted) Number of reads at the breakpoint position with an unaligned end, but weighted according to an alignment probability that is assigned to each read.
- Average quality Average read quality of a single position in the supporting unaligned ends at a breakpoint. Only reads that have unaligned ends with the same direction as the breakpoint (i.e left or right) are included in the average.
- Consensus sequence The consensus sequence calculated across the unaligned ends of the reads that support the breakpoint.
- Length Length of the consensus sequence.
- Complexity Complexity of the consensus sequence.