Perform QIAseq FastSelect RNA Expression and Fusion Calling (N6-T RT + ODT-RT primer)
The Perform QIAseq FastSelect RNA Expression and Fusion Calling (N6-T RT + ODT-RT primer) template workflow supports a combined N6-T RT primer + ODT-RT primer protocol. All reads are used for gene expression analysis while the reads amplified by the N6-T RT primer are used for studying transcript expression and fusions.
Figure 12.2 shows how the data is analyzed in two different arms of the workflow. A trim adapter list is used to separate the ODT-RT primer reads from the N6-T RT primer reads.
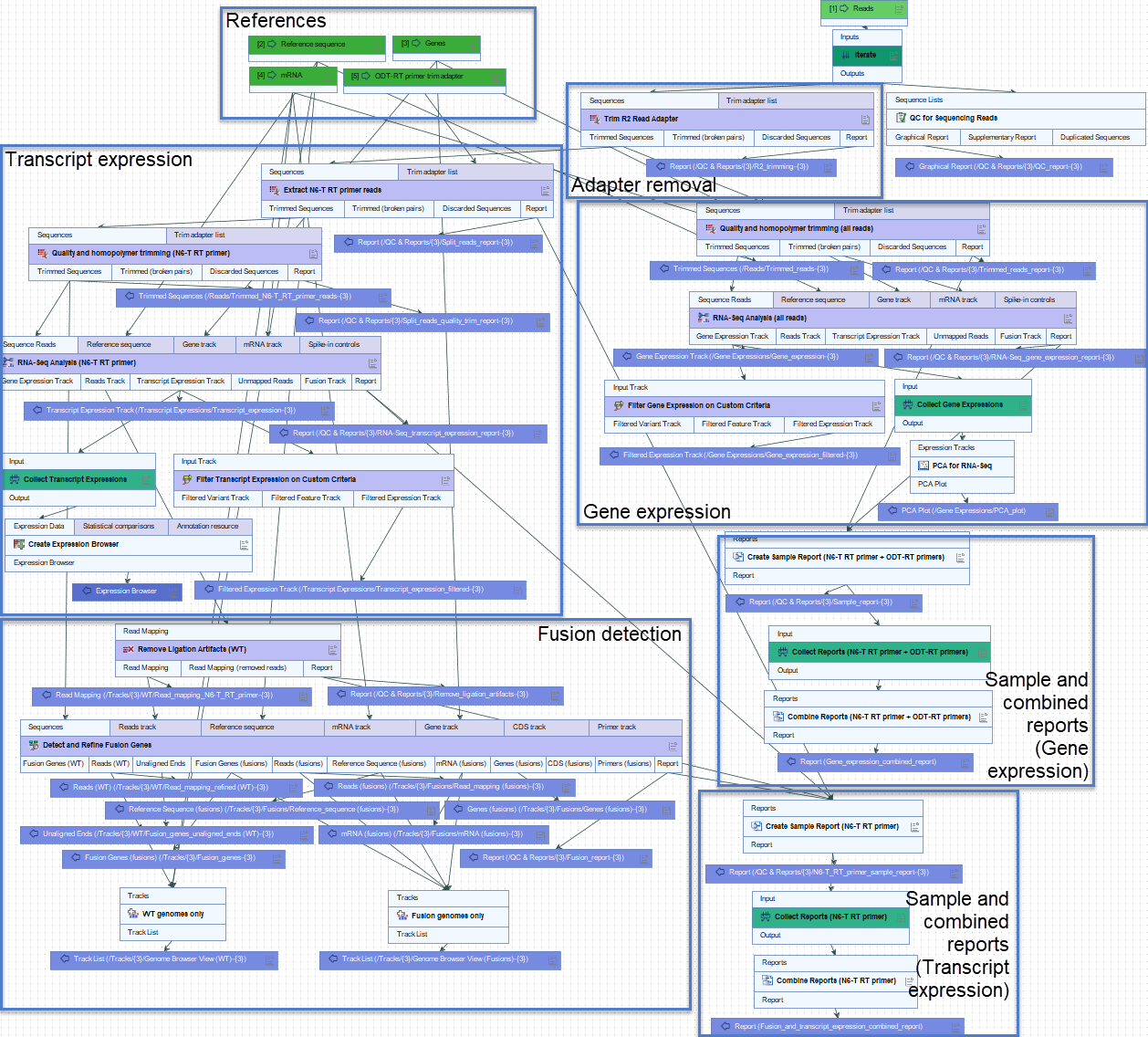
Figure 12.2: The Perform QIAseq FastSelect RNA Expression and Fusion Calling (N6-T RT + ODT-RT primer) workflow for analyzing the dual capture protocol. Each blue box encapsulates a particular part of the workflow.
The Perform QIAseq FastSelect RNA Expression and Fusion Calling (N6-T RT + ODT-RT primer) workflow can be found here:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | QIAseq RNA Workflows (
) | QIAseq RNA Workflows (![]() ) | Perform QIAseq FastSelect RNA Expression and Fusion Calling (N6-T RT + ODT-RT primer) (
) | Perform QIAseq FastSelect RNA Expression and Fusion Calling (N6-T RT + ODT-RT primer) (![]() )
)
Or run directly from the Analyze QIAseq Samples guide:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | Analyze QIAseq Samples (
) | Analyze QIAseq Samples (![]() )
)
where it is available in the dropdown menu of the FastSelect RNA tab by setting analysis to N6-T RT primer + ODT-RT primer.
In the first wizard step either select the reads or choose Select files for import to import FASTQ files on-the-fly. Note that the FASTQ files need to be imported upfront for Analyze QIAseq Samples to work.
The workflow is designed to analyze all samples within an experiment without the need of batch functionality. It iterates through the samples and processes them separately, and collects and distributes all outputs at the end.
Choose the QIAseq UPXome and FastSelect RNA hg38 reference data when running the workflow standalone.
Keep the "Use organization of input samples" except when you want to add metadata information to the samples. Check that the batch overview looks as expected, especially when using on-the-fly import.
Save the results to a specified location.
Subsections