How to run the De Novo Assemble PacBio Reads tool
If your input is raw SMRT sequencing reads, you should start by running the Correct PacBio Reads (legacy) tool to correct the reads.
To start the assembly tool go to:
Toolbox | Legacy Tools (![]() ) | De Novo Assemble PacBio Reads (legacy) (
) | De Novo Assemble PacBio Reads (legacy) (![]() )
)
This will open a dialog where you can select sequences to assemble. If you already selected sequences in the Navigation Area, these will be shown in 'Selected Elements'. You can alter your choice of sequences to assemble by using the arrows to move sequences between the Navigation Area and the 'Selected Elements' box. You can also add sequence lists.
Click Next to set the parameters for the assembly. This will show a dialog similar to the one in figure 14.6.
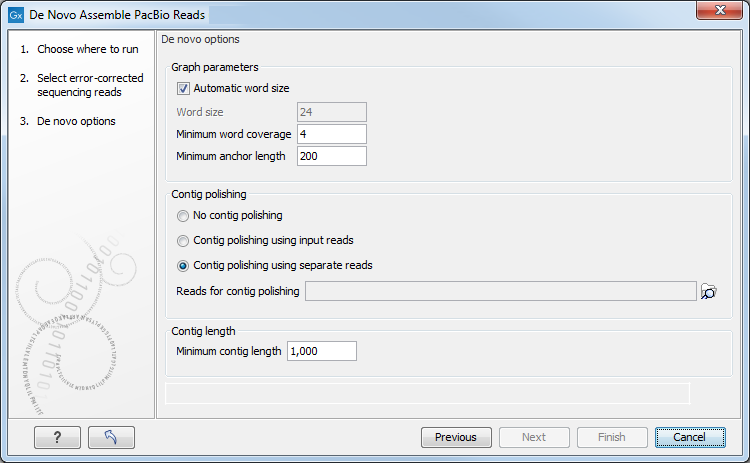
Figure 14.6: Select assembly parameters.
- Graph parameters
- Automatic word size The word size is automatically
estimated by default using the following formula14.1:
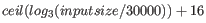 The word size can also be set manually. We recommend to use
a word size of 17-24. A small word size should be used for small
genomes, while a large word size should be used for large
genomes. When using an automatically estimated word size, you can see
the actual word size in the history (
The word size can also be set manually. We recommend to use
a word size of 17-24. A small word size should be used for small
genomes, while a large word size should be used for large
genomes. When using an automatically estimated word size, you can see
the actual word size in the history ( ) of
the result files. Please note that the range of word sizes is limited
to 12-64 on 64-bit machines.
) of
the result files. Please note that the range of word sizes is limited
to 12-64 on 64-bit machines.
- Minimum word coverage. It specifies the minimum number of times a given word must occur in the input reads in order for it to be included in the de Bruijn graph used by the assembler. The default minimum word coverage is 4. Using a smaller minimum word coverage will result in fewer contigs, while it may reduce the contig quality. Similarly, using a larger minimum word size will result in more contigs with a higher contig quality. If you have very high coverage, you may obtain a better assembly by choosing a larger minimum word coverage. Otherwise, we recommend that you leave it at 4.
- Minimum anchor length The minimum anchor length specifies the minimum length of anchor fragments that are retained in the assembly graph. The higher the value, the more noisy structure is removed from the graph. On the flip side, a too high setting can prevent complex stretches of the genome from being resolved by the assembler.
- Automatic word size The word size is automatically
estimated by default using the following formula14.1:
- Contig polishing Contig polishing is the last step of the assembly
algorithm, in which putative assembly errors in the contigs are
resolved by mapping a set of reads to the contigs and building a
consensus of this read mapping.
- No contig polishing will speed up the assembly process
- Contig polishing using input reads uses the error-corrected input reads that were used for the actual assembly
- Contig polishing using seperate reads uses another set of reads
- Minimum contig length Contigs below the specified length will not be reported. The default value is 1,000 bp. For very large assemblies, the number of contigs can be large, in which case the contig polishing-step will be slow. In this case, it is an advantage to raise the minimum contig length to reduce the number of contigs that have to be considered.
Click Next to set the output options, and finally click Finish to start the assembler.
Footnotes
- ... formula14.1
- The formula used in the regular assembler is
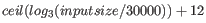 .
.