How to run the Join Contigs tool
Join Contigs can be launched from:
Tools | Genome Finishing Module (![]() ) |
Join Contigs (
) |
Join Contigs (![]() )
)
This opens the dialog shown in figure 11.2. Select the input contigs and click Next.
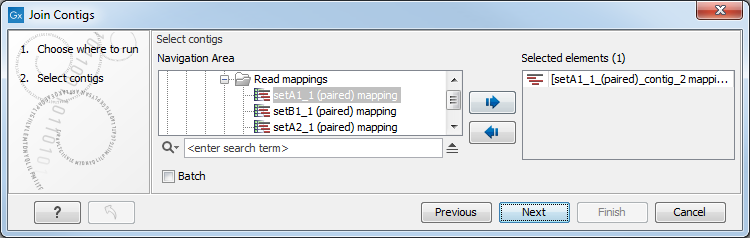
Figure 11.1: Select the contigs to use for joining.
The next dialog (shown in figure 11.2), contains options related to the four different types of analyses the tool can perform:
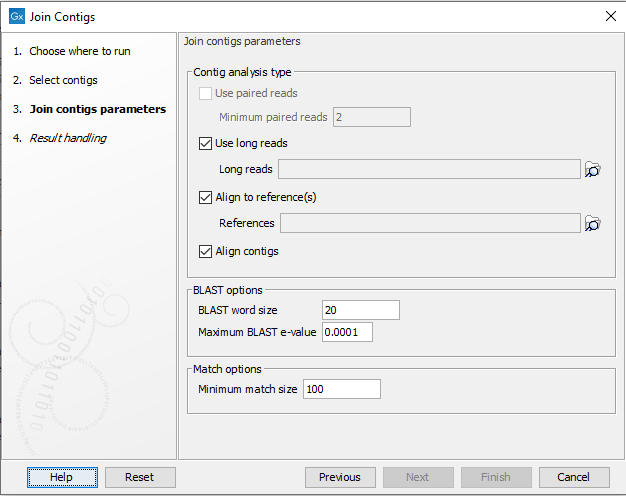
Figure 11.2: Options for detection of possible joins.
- Contig analysis types
- Use paired reads. When this option is selected, paired reads mapped to the contigs are used to detect neighboring contigs. "Minimum paired reads" is the minimum number of paired reads required to span two contigs before a join is considered.
- Use long reads. Enable the use of long reads for joining contigs. Click on the folder (
 ) to select one or more sets of long reads.
) to select one or more sets of long reads.
- Align to reference(s). Align the contigs to one or more reference sequences using BLAST and identify neighboring contigs. Click on the folder () to select the relevant reference(s).
- Align contigs. Align the contigs using BLAST and look for overlaps between contig ends.
- BLAST options BLAST is used to align contigs against reference sequences and for aligning contigs against each other.
- BLAST word size. Specifies the minimum number of nucleotides that must have a perfect alignment before BLAST finds a match. A small value increases the sensitivity but will result in more random matches and slow down the BLAST search on large data sets.
- Maximum BLAST e-value. The BLAST e-value indicates the number of hits that are expected by chance where an e-value of 0 indicate a unique hit while an e-value of 10 is a random match. Lowering the e-value threshold gives a more stringent alignment which help avoid misassemblies but it also decreases the chance of identifying neighboring contigs that can be joined.
- Match options
- Minimum match size. Specifies the minimum match size allowed in alignments.
When contigs are aligned against each other, the most interesting matches are often small overlaps between contig ends. To avoid that such small overlaps are filtered out due to a low e-value or minimum match size, contig ends are aligned in a separate step. The alignment of contigs ends allow matches of length ![]() 8bp and matches that are close to the contig ends are considered to be more significant compared to matches far from the contigs ends.
8bp and matches that are close to the contig ends are considered to be more significant compared to matches far from the contigs ends.
When it is possible to perform more than one of the four types of analyses described above, it is often a good idea to start out by performing each analysis separately. This will give an indication of how much each analysis contribute to improvements in the assembly. An analysis that cannot improve the assembly significantly on it own, will usually contaminate the graph build by the Join Contigs tool with bad information and thus make it hard to identify the correct joins. For example, if both long reads and a reference sequence is available, then running the Join Contigs tool with both can result in an inferior result compared to just using the long reads. This usually happens when the reference sequence is contain too many structural variations compared to the organism which was sequenced. In other words, the reference and the long reads will not agree on the set of possible joins.
In the Result handling step (shown in figure 11.3), specify which tables to output before clicking Finish.
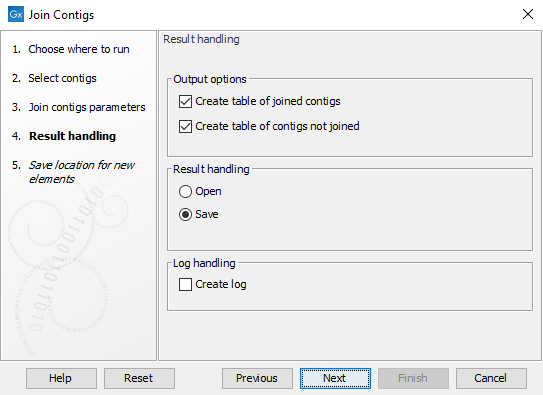
Figure 11.3: Specify which tables to output with details of the join process.
The tool proposes the creation of two output table. The primary output is a table of joined contigs. It lists all contigs that are resulting from a join between two or more input contigs, as well as details about the join itself (figure 11.4).
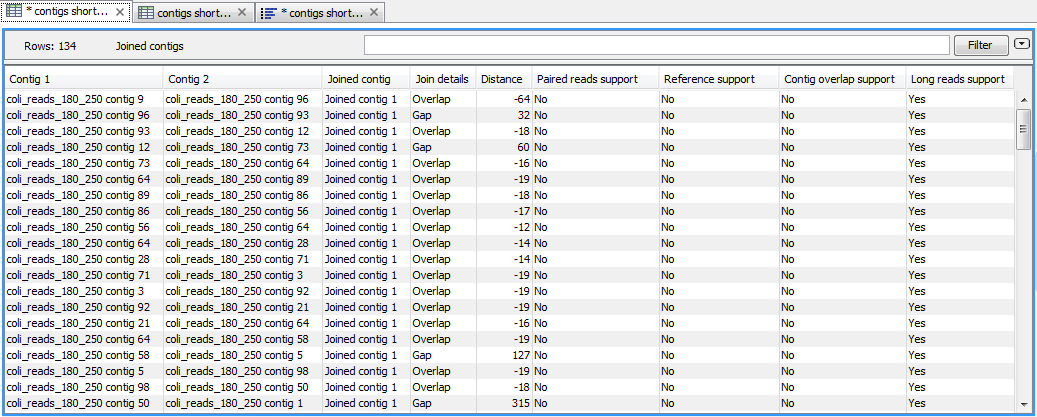
Figure 11.4: Table containing details on each join made by the tool.
An annotation on the sequence also indicates whether the join was performed using an overlap or a gap (figure 11.5).
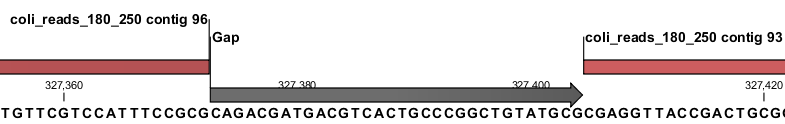
Figure 11.5: An example of a gap between two contigs that has been filled based on long reads.
The second output is a table of contigs not joined (see figure 11.6). The column 'Reason' differentiates between two sorts of contigs:
- 'Not part of any join' describes contigs that were not joined at all. It can happen if the contigs are a result of contamination in the sample or if there was insufficient information to join the contig correctly.
- 'Repeat not included enough times' are contigs that were identified as repetitive and joined in some contigs, but not in all the contigs expected based on the estimated copy number of the repeat contig calculated by the Contig Joiner tool.
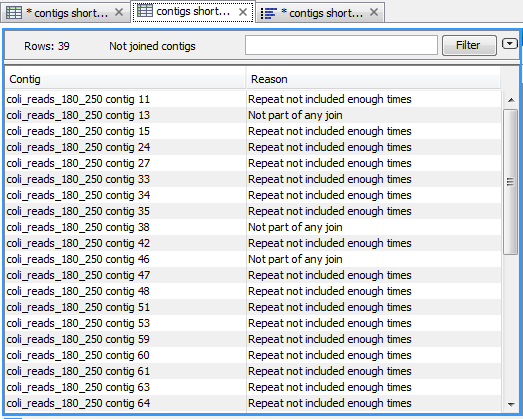
Figure 11.6: Table containing a list of contigs that was not part of any join, or not part of enough joins in the case of repeat contigs.