Join Contigs
The Join Contigs tool provides an automated way of joining contigs based on the following types of analyses:
- Long reads, such as PacBio reads, can be used to join contigs if they span more than one contig. Long reads are mapped to the contigs iteratively using the CLC read mapper by using unmapped regions of reads from one iteration as input reads to the following iteration. If the tool estimates that two contigs should be joined with a gap in between, an attempt is made to fill the gap using an alignment of the reads spanning the gap. If the quality of this read alignment is too low, the gap is filled with N's instead. A weight is computed for each possible join based on how well the reads map to the two contigs.
- Paired reads that span multiple contigs are used to identify possible neighboring contigs which can be joined. The Join Contigs tool only consider reads that map close to the contig ends to prevent spurious matches from repeat regions embedded in the contigs. Through the Join Contigs wizard, it is possible to specify a minimum number of paired reads that must span two contigs before they are considered in a join. A weight is computed for each possible join based on the number of paired reads spanning the two contigs and the standard deviation of the distance estimate as follows:
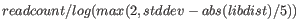
where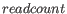 is the number of paired reads supporting the join,
is the number of paired reads supporting the join, 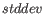 is the standard deviation and
is the standard deviation and 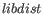 is the expected paired library distance.
is the expected paired library distance.
- An alignment of the contigs to a closely related reference. Contigs are first aligned to the reference using the Align Contigs tool. Next spurious matches are filtered as follows.
- Matches which only cover a small fraction of contigs are ignored.
- Overlapping matches are evaluated with respect to the match size and the identity if the match. If one match is significantly larger than the other match or has significantly higher identity, we ignore the smallest or lowest identity match if
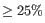 of this match is overlapped by the other match.
of this match is overlapped by the other match.
- Overlapping contigs are detected by aligning contigs against each other using the Align Contigs tool. A weight for each possible join is computed based on the number of mismatches in the overlapping region and the position of the overlap. Overlaps close to the edge of a contig give rise to higher weights than an overlap located in the middle of a contig.
The Join Contigs tool builds a graph over all possible joins based on the four analyses above where edges represents possible joins and nodes represent contigs. Each edge is assigned a weight as described above. If a join is ambiguous, i.e. two or more analyses disagree on a join, one of the following events can happen:
- The weights of two or more joins are within the same range. In this case nothing is done.
- The weight for one join is significantly higher than the weights for all alternative joins. The join with the highest weight is performed.
Subsections