Joining two contigs
It can be relevant to join two contigs for several reasons - e.g. if you:
- detect two overlapping contigs using the contig aligner.
- have contigs which map to the reference genome and are separated by a gap.
- have resequenced regions, made de novo assembly with the resequenced reads included and want to join the new contigs with the existing ones.
It is possible to join two contigs in different ways.
- Joining contigs using the Join Contigs button in the Contig table view (figure 2.7) is performed without using a reference sequence. You can select the two contigs you wish to join in the Contig table by holding down the ctrl-key and clicking on the two contigs. Alternatively you can select two contigs from the Contig match view and then select a region in the reference containing matches from the two contigs. Because the Contig match view is synchronized with the Contig table, contigs in the selected region will be selected in the match table.
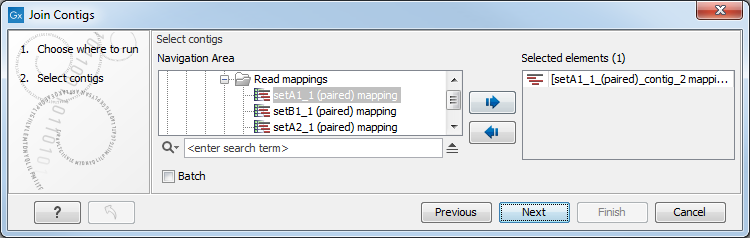
Figure 2.7: Contig Table - Join contigs wizardIn both case, clicking the Join Contigs button opens a wizard with the following options:
- Automatic find overlap and align: A function that identifies the overlap between two contigs using BLAST followed by an alignment to calculate the consensus contig. This function favors overlaps at the ends of the contigs.
- Manual gap: Function that can be used to join sequential and non-overlapping contigs when the orientation and gap size is known. When ticked, gap size and contig orientation must be specified.
- It is also possible to join two contigs from the Contig match view by selecting a region in the reference sequence where two contigs overlap and right click that selection. Select Join Two Contigs from the drop down menu (figure 2.8) and specify the contigs to be joined in the dialog window. The wizard lists all contig matches in the selected region and the contigs to use in the join are selected by selecting the corresponding matches.
- Select first contig match. Select the first contig match from the list to use for the join.
- Select second contig match. Select the second contig match from the list to use for the join.
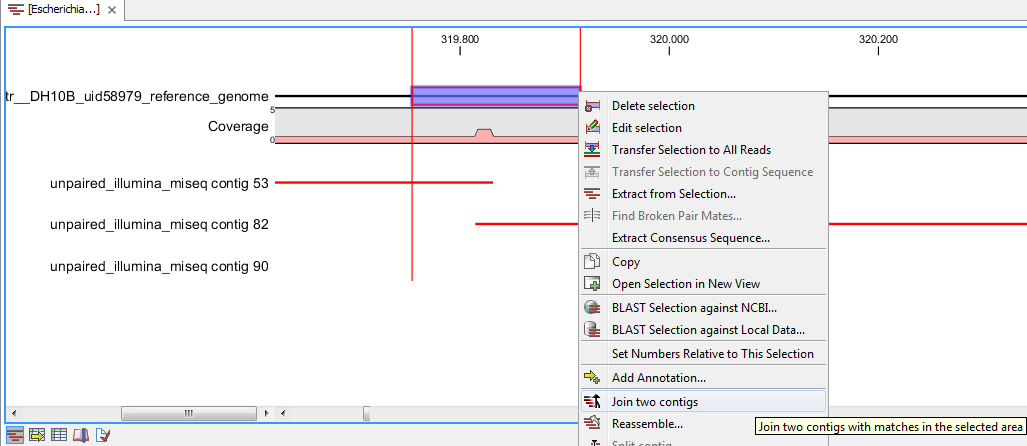
Figure 2.8: Match view - Join Contigs wizardThis method is very useful in cases where an overlap between two contigs is very short. This method only considers overlaps that are present in the selection made. The automatic join method, described earlier, would fail to consider a short overlap, favoring other more significant ones instead. With this method, you have control over the location of the overlap, making it possible to join contigs that overlap by as little as a single nucleotide.
For all join methods described above it is possible to keep the old contigs. This is done by ticking Keep contig under Old contigs, which is useful when joining contigs that represent repetitive elements needed for joining other contigs elsewhere in the mapping.
Note! When joining two contigs, the orientation of the result is not guaranteed to follow the orientation of the original contigs, e.g. two contigs with reverse orientation relative to the reference can result in a contig with forward orientation depending on the join function used. However, the orientation of contigs is usually of no importance and the CLC de novo assemblers will output contigs with a somewhat arbitrary orientation.