How to run the Collect Paired Read Statistics tool
To launch Collect Paired Read Statistics, go to:
Tools | Genome Finishing Module (![]() ) |
Collect Paired Read Statistics (
) |
Collect Paired Read Statistics (![]() )
)
This opens the dialog shown in figure 8.1.
Figure 8.1: Select the read mappings to analyze.
Select the relevant read mappings and click Next. The next wizard window ( figure 8.2) makes it possible to choose how the paired reads statistics are collected. The default option is to only consider reads that map to the contig ends which help filter out noise from reads that are erroneously mapped or reads that map to repetitive regions and thus make it easier to determine if two contigs are neighbors. Alternatively, statistics can be generated from all read pairs mapped to the contigs, which can make misassemblies evident as large overlaps between contigs. It is also possible to restrict collection of paired statistics to reads from specific paired libraries. This is done in step 2 of the wizard by selecting the option Include subset of libraries and then selecting one or more libraries which have reads mapped to the contigs. Please note that the libraries are named after the file from which the reads were imported.
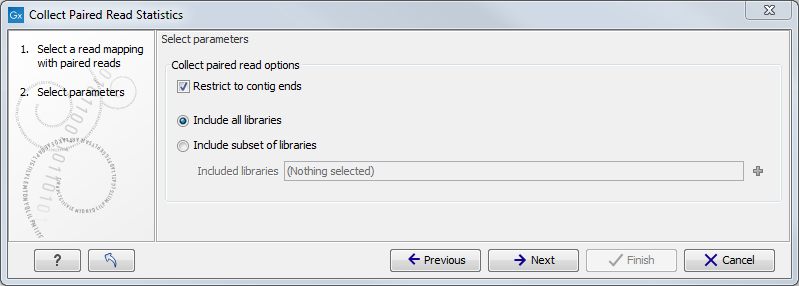
Figure 8.2: Select whether to collect paired reads only from the ends of contigs or from the entire contig. Optionally, restrict the collection of paired statistics to a subset of paired libraries.
Finally click Next and Finish.
Note: The Collect Paired Reads Statistics should only be performed on de novo assemblies where the contig has not been edited. If run on modified contigs, the distance estimates will not be accurate. If your contigs have been modified, you can extract the contig sequences by opening the de novo assembled data, select all contigs and click on Extract Contig. The extracted contig sequences can next be used as reference in a new read mapping using the NGS core tool Map Reads to Contigs. This new read mapping can now be used as input in the Collect Paired Read Statistics tool.