The annotated variant table
The annotated variant table (see figure 21.91) contains a subset of the columns of the variant track table and additionally the three columns below.
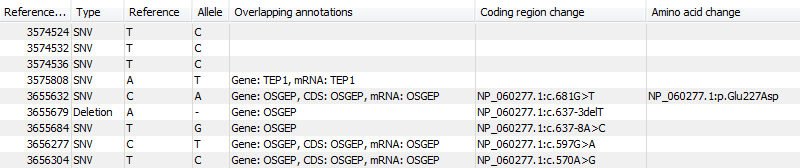
Figure 21.91: An example of an annotated variant table.
When the variant calling is performed on a read mapping in which gene and cds annotations are present on the reference sequence, the three columns will contain the following information:
- Overlapping annotation
- This shows if the variant is covered by an annotation. The annotation's type and name will displayed. For annotated reference sequences, this information can be used to tell if the variant is found in e.g. a coding or non-coding region of the genome. Note that annotations of type
VariationandSourceare not reported. - Coding region change
- For variants that fall within a coding region of a gene, the change is reported according to the standard conventions as outlined in http://www.hgvs.org/mutnomen/.
- Amino acid change
- If the reference sequence of the mapping is annotated with ORF or CDS annotations, the variant caller will also report whether the variant is synonymous or non-synonymous. If the variant changes the amino acid in the protein translation, the new amino acid will be reported. The nomenclature used for reporting is taken from http://www.hgvs.org/mutnomen/.
The table can be Exported (![]() ) as a csv file (comma-separated values) and imported into e.g. Excel. Note that the CSV export includes all the information in the table, regardless of filtering and what has been chosen in the Side Panel. If you only want to use a subset of the information, simply select and Copy (
) as a csv file (comma-separated values) and imported into e.g. Excel. Note that the CSV export includes all the information in the table, regardless of filtering and what has been chosen in the Side Panel. If you only want to use a subset of the information, simply select and Copy (![]() ) the information.
) the information.
Note that if you make a split view of the table and the mapping, you will be able to browse through the variants by clicking in the table. This will cause the view to jump to the position of the variant.
This table view is not well-suited for downstream analysis, in which case we recommend working with tracks instead (see Variant tracks).