Definition of wildcards
One of the automation configuration settings is the Sequencer folder as described in Sequencer folder. When a new sequencing run is started, the sequencer will output the results from the sequencing run to the sequencer folder (see example in figure 4.6).
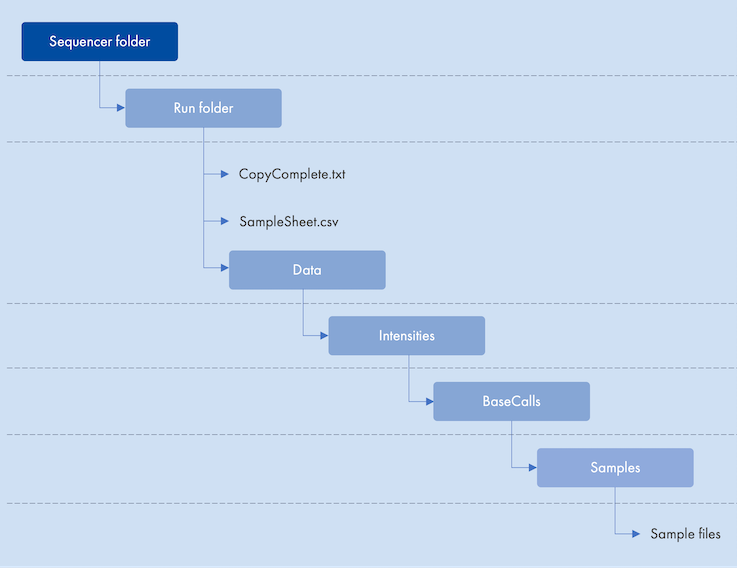
Figure 4.6: Example of output from a sequencer
The Workflow matcher, Sample completion, Sample sheet, and Batch folder settings point to objects inside the sequencer folder. When configuring these settings, the file system paths relative to the sequencer folder should be provided. Using the example in figure 4.6, the path to a sample sheet could look like this:
220522-000000_NDX1337_RUO_0001_XPNEKVSPRX/SampleSheet.csv
However, parts of this path contains information that changes from run to run, such as the ID of the sequencing run and timestamps. Any such parts should be replaced with wildcards when configuring the settings. Using the sample sheet path above as an example, the configured setting could look like this:
*_NDX*_RUO_*_*/SampleSheet.csv
It is recommended that wildcards are only used if necessary as excessive use of wildcards increases the risk of matching objects that were not intended to be matched. For example, if the sequencer produces sample files for two workflows, CAT and CATARACT, installed on the CLC Genomics Server, e.g. ID1234_CAT_S1_L001_R1_001.fastq.gz and ID5678_CATARACT_S2_L001_R1_001.fastq.gz, the following Workflow matcher setting for the CAT workflow will accidentally match the CATARACT sample files resulting in these samples inadvertently being submitted to the CAT workflow:
*_NDX*_RUO_*_*/Data/Intensities/BaseCalls/Samples/*CAT*
To prevent this from happening, the "_" surrounding CAT should be explicitly included in the Workflow matcher setting:
*_NDX*_RUO_*_*/Data/Intensities/BaseCalls/Samples/*_CAT_*