Trim output
Clicking Next will allow you to specify the output of the trimming as shown in figure 21.14.
Figure 21.14: Specifying the trim output.
In most case, independently of what option are selected in this dialog, a list of trimmed reads will be generated:
- Sequence elements (individual sequences) selected as input and not discarded during trimming will be output into a single sequence list, as long as one or more of the input sequences were trimmed.
- Sequence lists selected as input will be output as as many corresponding sequence list, assuming that at least one sequence in any one of the sequence lists input was trimmed.
However, if no sequences are trimmed using the parameter settings provided, then no sequence lists are output when running the tool directly. A warning message appears stating that no sequences were trimmed. When the tool is run within a workflow, and if no sequences are trimmed using the parameter settings provided, then all input sequences are passed to the next step of the analysis via the "Trimmed Sequences" output channel.
In addition the following can be output as well:
- Save discarded sequences. This will produce a list of reads that have been discarded during trimming. Sections trimmed from reads that are not themselves discarded will not appear in this list.
- Save broken pairs. This will produce a list of orphan reads.
- Create report. An example of a trim report is shown in figure 21.15. The report includes the following:
- Trim summary.
- Name. The name of the sequence list used as input.
- Number of reads. Number of reads in the input file.
- Avg. length. Average length of the reads in the input file.
- Number of reads after trim. The number of reads retained after trimming. This includes both paired and orphan reads.
- Percentage trimmed. The percentage of the input reads that are retained.
- Avg. length after trim. The average length of the retained sequences.
- Read length before / after trimming. This is a graph showing the number of reads of various lengths. The numbers before and after are overlayed so that you can easily see how the trimming has affected the read lengths (right-click the graph to open it in a new view).
- Trim settings A summary of the settings used for trimming.
- Detailed trim results. A table with one row for each type of trimming:
- Input reads. The number of reads used as input. Since the trimming is done sequentially, the number of retained reads from the first type of trim is also the number of input reads for the next type of trimming.
- No trim. The number of reads that have been retained, unaffected by the trimming.
- Trimmed. The number of reads that have been partly trimmed. This number plus the number from No trim is the total number of retained reads.
- Nothing left or discarded. The number of reads that have been discarded either because the full read was trimmed off or because they did not pass the length trim (e.g. too short) or adapter trim (e.g. if Discard when not found was chosen for the adapter trimming).
- Automatic adapter read-through trimming. This section contains statistics about how many reads were automatically trimmed for adapter read-through. It will also list the two detected read-through sequences.
- Trim summary.
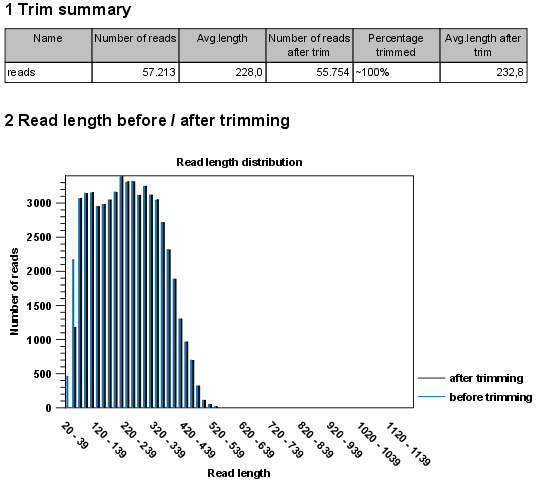
Figure 21.15: A report with statistics on the trim results. Note that the Average length after trimming (232,8bp) is bigger than before trimming (228bp) because 2.000 very short reads were discarded in the trimming process.
If you trim paired data, the result will be a bit special. In the case where one part of a paired read has been trimmed off completely, you no longer have a valid paired read in your sequence list. In order to use paired information when doing assembly and mapping, the Workbench therefore creates two separate sequence lists: one for the pairs that are intact, and one for the single reads where one part of the pair has been deleted. When running assembly and mapping, simply select both of these sequence lists as input, and the Workbench will automatically recognize that one has paired reads and the other has single reads.