The Identify QIAseq DNA Variants template workflows
The Identify QIAseq DNA Variants template workflows are optimized to work with either somatic or germline applications from Illumina or Ion Torrent reads.
Two different types of panels are available for QIAseq Targeted DNA analysis, QIAseq Targeted DNA panels and QIAseq Targeted DNA Pro panels. The read structure is different between the two types of panels, and it is therefore important to choose the correct workflow to allow proper trimming and UMI grouping of the reads. Panel IDs for QIAseq Targeted DNA applications start with DHS or CDHS whereas panel IDs for QIAseq Targeted DNA Pro applications start with PHS or CPHS.
The workflows handling the two types of QIAseq panels are very similar, but default tool settings and the order of tools in the variant filtering cascades differ.
- General differences between QIAseq DNA and QIAseq DNA Pro analysis workflows:
- A number of settings in the two tools Remove and Annotate with Unique Molecular Index and Trim reads differ, as they have been set up to handle reads from the relevant type of QIAseq panel appropriately.
- In Pro workflows, an additional base after the primer is unaligned.
- Differences between Illumina QIAseq DNA and Illumina QIAseq DNA Pro analysis workflows:
- In QIAseq DNA workflows, the minimum read length after trimming is set to 20. This has been increased to 40 in the QIAseq DNA Pro workflows.
- The filtering cascades used for germline variant filtering varies widely between QIAseq DNA and QIAseq DNA Pro analysis workflows. Whereas the QIAseq DNA workflow has an extensive series of filtering steps, the QIAseq DNA Pro workflow has a relatively simple filtering cascade.
- Differences between Ion Torrent QIAseq DNA and Ion Torrent QIAseq DNA Pro analysis workflows:
- In QIAseq DNA workflows, the mismatch cost in Map Reads to References is 2. This has been increased to 4 in the QIAseq DNA Pro workflows.
- In QIAseq DNA workflows, the Minimum supporting consensus fraction in Create UMI Reads from Grouped Reads is 0.0. This has been increased to 0.5 in the QIAseq DNA Pro workflows.
- In QIAseq DNA workflows, the variant frequency in Remove False Positives is 0.5. This has been increased to 2 in the QIAseq DNA Pro workflows.
In the following, Identify QIAseq DNA and Identify QIAseq DNA Pro workflows are described together and are only mentioned specifically when there is a relevant difference.
To support QIAseq Targeted DNA analysis, the following workflows are available:
- Identify QIAseq DNA Somatic Variants (Illumina)
- Identify QIAseq DNA Somatic Variants (Ion Torrent)
- Identify QIAseq DNA Germline Variants (Illumina)
- Identify QIAseq DNA Germline Variants (Ion Torrent)
- Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina)
To support QIAseq Targeted DNA Pro analysis, the following workflows are available:
- Identify QIAseq DNA Pro Somatic Variants (Illumina)
- Identify QIAseq DNA Pro Somatic Variants (Ion Torrent)
- Identify QIAseq DNA Pro Germline Variants (Illumina)
Note that the Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina) differs from the other QIAseq DNA workflows by calling both somatic and germline variants in the same workflow and is described separately in Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina).
Somatic/germline specificity: For somatic variant detection, the template workflow uses the Low Frequency Variant Detection tool, a variant caller that does not base its statistical model on a bi-allelic assumption. This variant caller will thus declare a site heterozygous if it detects more than one allele at that site, even if one of the alleles is detected at very low frequency and later filtered out. For germline applications, the workflows use the Fixed Ploidy Variant Detection tool. This variant caller has higher precision than the Low frequency Variant Detection tool, particularly at low to moderate levels of coverage (< 30x). At high levels of coverage (>100x) the Fixed Ploidy Variant Detection tool will exhibit low sensitivity for variants with allele frequencies far from what is expected for germline variants (that is 50 or 100%). For more information about the variant callers, please see: http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Low_Frequency_Variant_Detection.html and http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Fixed_Ploidy_Variant_Detection.html.
Illumina/Ion Torrent specificity: Among various differences in the filtering strategy applied in the workflows aimed at analyzing data from a particular sequencing technology, the workflow for Ion Torrent data includes an extra step that removes non SNV type variants that are likely due to artifacts.
In each case, the parameter values applied as defaults have been optimized for high sensitivity and specificity when detecting variants.
The following description applies to the Identify QIAseq DNA (Pro) Variants template workflows optimized for calling either somatic or germline variants:
The QIAseq DNA workflows use the Reference Data set QIAseq DNA Panels hg19 whereas the QIAseq DNA Pro workflows use QIAseq DNA Pro Panels hg38. Before starting one of the workflows for the first time, open the Reference Data Manager and select and download the relevant reference data set if you have not already done so.
The Identify QIAseq DNA Variants template workflows can be found here:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | QIAseq Analysis workflows (
) | QIAseq Analysis workflows (![]() ) | Identify QIAseq DNA Somatic/Germline Variants (Illumina/Ion Torrent) (
) | Identify QIAseq DNA Somatic/Germline Variants (Illumina/Ion Torrent) (![]() )
)
And the Identify QIAseq DNA Pro Variants template workflows can be found here:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | QIAseq Analysis workflows (
) | QIAseq Analysis workflows (![]() ) | Identify QIAseq DNA Pro Somatic/Germline Variants (Illumina/Ion Torrent) (
) | Identify QIAseq DNA Pro Somatic/Germline Variants (Illumina/Ion Torrent) (![]() )
)
Double-click on the relevant workflow to run the analysis.
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the Select reads dialog, specify the sequencing reads that should be analyzed (figure 6.1).
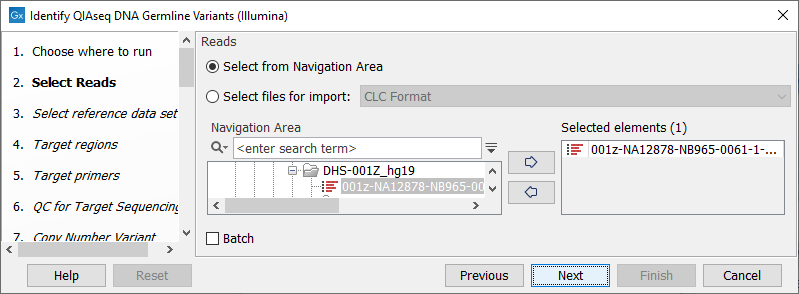
Figure 6.1: Select the sequencing reads by double-clicking on the file name or by clicking once on the file name and then on the arrow pointing to the right hand side.
The following dialog helps you set up the relevant Reference Data Set. If you have not downloaded the Reference Data Set yet, the dialog will suggest the relevant data set and offer the opportunity to download it using the Download to Workbench button. This is shown in (figure 6.2).
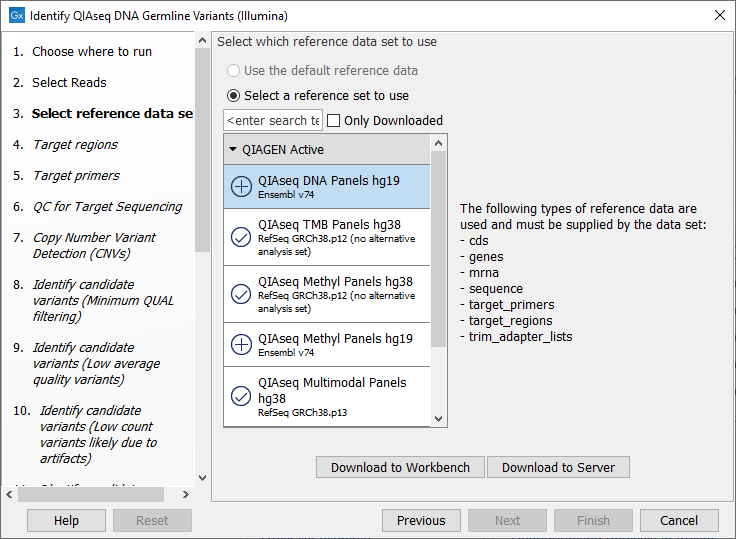
Figure 6.2: The relevant Reference Data Set is highlighted; the types of reference needed by the workflow are listed in the text to the right. For QIAseq Targeted DNA workflows QIAseq DNA Panels hg19 will be highlighted, whereas for QIAseq Targeted DNA Pro workflows, QIAseq DNA Pro Panels hg38 will be highlighted.
Note that if you wish to Cancel or Resume the Download, you can close the template workflow and open the Reference Data Manager where the Cancel, Pause and Resume buttons are available.
If the Reference Data Set was previously downloaded, the option "Use the default reference data" is available and will ensure the relevant data set is used. You can always check the "Select a reference set to use" option to be able to specify another Reference Data Set than the one suggested.
In the next dialog (figure 6.3), specify the relevant target regions from the drop down list.
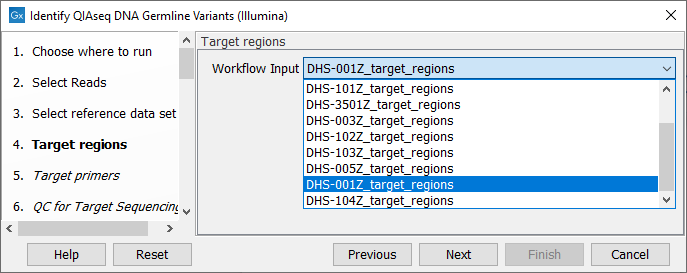
Figure 6.3: Select the target regions file specific to the panel used.
In the next dialog (figure 6.4), specify the relevant target primers from the drop down list.
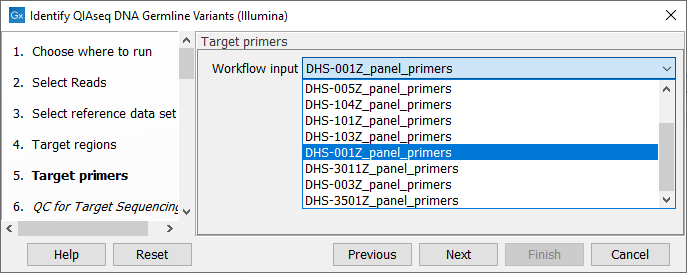
Figure 6.4: Select the target primers file specific to the panel used.
In the dialog called QC for Target Sequencing, you can modify the Minimum coverage needed on all positions in a target for this target to be considered covered (figure 6.5). Note that the default value for this tool depends on the application chosen (somatic or germline).
Figure 6.5: Setting the Minimum coverage parameter of the QC for Target Sequencing.
The dialog for Copy Number Variant Detection allows you to specify a control mapping against which the coverage pattern in your sample will be compared in order to call CNVs. If you do not specify a control mapping, or if the target regions files contains fewer than 50 regions, the Copy Number Variation analysis will not be carried out.
Please note that if you want the copy number variation analysis to be done, it is important that the control mapping supplied is a meaningful control for the sample being analyzed. Mapping of control samples for the CNV analysis can be done using the workflows described in Create QIAseq DNA CNV Control Mapping workflows.
A meaningful control must satisfy two conditions: (1) It must have a copy number status that it is meaningful for you to compare your sample against. For panels with targets on the X and Y chromosomes, the control and sample should be matched for gender. (2) The control read mapping must result from the same type of processing that will be applied to the sample. One way to achieve this is to process the control using the workflow (without providing a control mapping for the CNV detection component) and then to use the resulting UMI reads track as the control in subsequent workflow runs.
If you have previously run the workflow with control data, you will find the mapping in the Reports and Data folder (Mapped UMI Reads).
The parameters for variant detection are not adjustable and have been set to generate an initial pool of all potential variants. These are then passed through a series of filters to remove variants that are suspected artifacts. Variants failing to meet the (adjustable) thresholds for quality, read direction bias, location (low frequency indels within homopolymer stretches), frequency or coverage would not be included in the filtered output.
Some filters only remove alternative alleles - and not reference alleles - as this potentially leads to wrong interpretation of variants by the VCF exporter where such variants could be misinterpreted as hemizygote when the reference allele is missing.
Note that each filter has been configured with specific default values depending on technology (Illumina / Ion Torrent), application (somatic or germline) and panel type (Targeted DNA/Targeted DNA Pro) chosen to provide the best sensitivity and precision in the variants output by each workflow. However, benchmarking was performed on samples of relatively high coverage. Therefore, additional filtering might be needed, or filtering values adjusted when working with low coverage samples. This can only be done by running the workflows listed in the Toolbox, and not by using the Analyze QIAseq Samples guide. When configuring filters, do not load any annotations, nor try to change the name of the filters in the first column, as it would disable the filter completely.
Note that reads that span the origin of the MT chromosome are not trimmed by the Trim Primers of Mapped Reads tool when running the Identify QIAseq DNA Variants template workflows on data from the DHS-105Z panel.
Subsections
- Output from the Identify QIAseq DNA Variants workflows
- Quality Control for the Identify QIAseq DNA Variants workflow
- Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina)
- Output from the Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina) workflow