Import Immune Reference Segments
The Import Immune Reference Segments tool can import reference sequences for V, J and C segments from a fasta file. The sequences are needed when running "Immune Repertoire Analysis" for either T or B cell receptor repertoire (TCR and BCR, respectively) (see Immune Repertoire Analysis).
The importer can be found here:
Import (![]() ) | Import Immune Reference Segments (
) | Import Immune Reference Segments (![]() ).
).
The importer can be used to import fasta files that are either in the IMSEQ [Kuchenbecker et al., 2015] or IMGT [Lefranc et al., 2009] format (see figure 8.4).
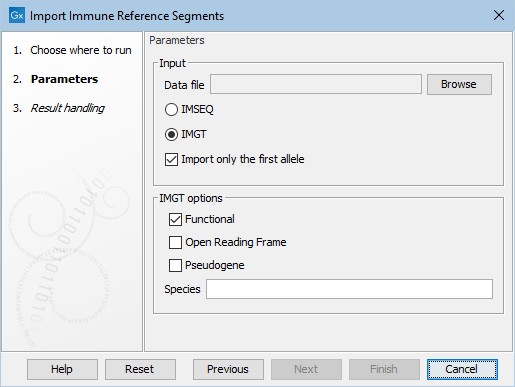
Figure 8.4: The available options when importing immune reference segments.
Both formats support allele numbering for the gene segments. If Import only the first allele is ticked, only segments without an allele or those with an allele defined as the number "1" (i.e "01" is also valid) will be imported. Otherwise, all segments are imported.
The two formats differ in how the sequence header is parsed for identifying the gene segment and related information, and how the conserved amino acids in the V and J segments are identified.
When saving the results, the reference data for either TCR, or BCR, or both, can be saved. The wizard will show an error message if an output option is ticked for which no relevant reference sequences are available.
The importer can only handle one fasta file at a time, but if two or more fasta files are imported, the resulting sequence lists can subsequently be combined to one list using the Create Sequence List tool.
IMSEQ
For the IMSEQ format, the header contains the following elements, separated by "|":
- The chain: TRA, TRB, TRG or TRD for T cells, and IGH, IGK and IGL for B cells.
- The segment type: V, J. Constant genes for T cells have the type C, while for B cells the constant genes are designated by the letter (and optionally number) corresponding to the encoded isotype.
- The segment ID.
- The segment allele.
- For J and V segments, the position of the first base of the conserved amino acid, counting from 0.
Currently only the heavy (IGH) and light ![]() and
and ![]() (IGK and IGL) chain types are supported for B cells.
(IGK and IGL) chain types are supported for B cells.
Any segments with an unsupported chain or segment type are silently ignored.
IMGT
For the IMGT format, the header contains 15 elements, separated by "|". Only the following are read and used during import:
- (1) Accession number(s).
- (2) The segment name, including chain, segment type, ID and allele, in the format: <chain><type><ID>*<allele>, for example "TRAV1*01".
Chain and segment type are the same as for IMSEQ.
- (3) Species.
- (4) Allele functionality: F (functional), P (pseudogene) or ORF (open reading frame).
- (5) Extracted label(s): EX1 and CH1 for C segments, and J-REGION and V-REGION for J and V segments, respectively.
- (8) The start of the codon, counting from 1, or "NR" for non coding labels.
- (9) The number of nucleotides added in 5' in the format +n.
The IMGT database contains chains, segment types and labels that are not listed above and are not supported. These are silently ignored.
|
While the IMSEQ format provides the position of the conserved amino acid, this needs to be calculated for the IMGT format. For this, the V region needs to be provided with gaps such that the conserved amino acid is found at approximately position 104 in the translated amino acid sequence. When downloading sequences from the IMGT database in fasta format, the "F+ORF+in-frame P nucleotide sequences with IMGT gaps" should be used. Alternatively, the corresponding "nt-WithGaps-F+ORF+inframeP" flat file can be downloaded from IMGT/GENE-DB.
If using custom reference data that is not downloaded from the IMGT database, it is recommended to use the IMSEQ format and specify the position of the conserved amino acid. |
When importing files in the IMGT format, the following options are available (see figure 8.4):
- Which allele functionality(ies) should be imported. At least one must be chosen.
- Which species should be imported. After choosing the fasta file, the desired species can be chosen from the list of species identified in the file.
If element (9) in the header is not empty, the corresponding number of nucleotides are removed from the 5' end of the sequence.
Identification of the conserved amino acid
The nucleotide sequence (with IMGT gaps for the V segments), starting from position in element (8) in the header, is first translated to amino acids using the standard genetic code. The position of the conserved amino acid is calculated, and, if identified, translated to the position of the first nucleotide in the corresponding codon. Segments where the amino acid cannot be identified are silently ignored.
For the V segments, the amino acid position is calculated as follows:
- If the amino acid at position 104 is C, then position 104 is used.
- Otherwise, the position of the last occurrence of C after position 104 is used, if present.
- Otherwise, if the amino acid at position 104, 105 or 103 (in this order) is one base pair mutation away from C and not a stop codon (i.e. R, S, C, F, G, W, Y), then this position is used.
For the J segments, all 3 open reading frames (starting from nucleotide position 1, 2 or 3) are used. Note that "." below denotes any amino acid. The amino acid position is calculated as follows:
- The amino acid sequence "(F|W)G.G", if present, is identified.
- The open reading frame that contains the amino acid sequence, no stop codon and has the lowest nucleotide starting position, if any, is used.
- Otherwise, the open reading frame that contains the amino acid sequence and at least one stop codon, if any, is used. If multiple open reading frames match this criteria, none are used.
- Otherwise, the amino acid sequences "(F|W)X.G" and "(F|W)G.X", if present, are identified. Here, X denotes the amino acids that are one base pair mutation away from F/W and not a stop codon (i.e. A, R, S, C, D, E, V, W).
- For each of the two amino acid sequences, the position is calculated as above.
- If both amino acid sequences are present, the position that is closest to the end of the sequence is used.
V and J segments for which the amino acid position cannot be successfully identified are silently ignored.
Output from Import Immune Reference Segments
The importer outputs a trim adapter list and sequence list that can be used for immune repertoire analysis. These can be added to a custom reference data set, to be used in workflows. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Custom_Sets.html for details.
The trim adapter list contains the first 21bp of the C segments found in the fasta file, and it is configured to trim the 5' end of the reads (see figure 8.5). For protocols where the C segment is at the 3' end of the read, the trim adapter list can be manually updated, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Creating_new_Trim_adapter_list.html.
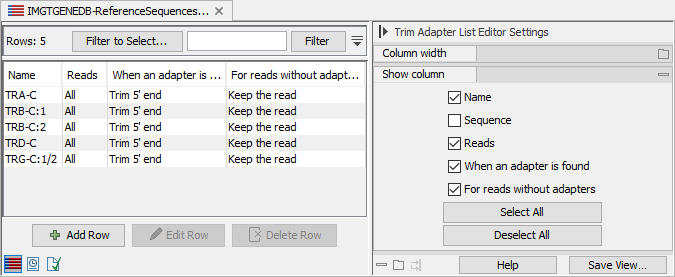
Figure 8.5: Imported trim adapter list showing the names and actions to be performed when the adapters are found or not. TRG-C-1 and TRG-C-2 share their first 21bp and jointly lead to the "TRG-C:1/2" adapter.
The sequence list contains the reference sequences for the V and J segments, named in the format <chain>-<type>-<ID>*<allele>, for example "TRA-V-1*01". If the gene segment does not have an allele or Import only the first allele is ticked, the *<allele> is not added to the name.
By ticking "Show annotations" and "Region" in the Side Panel "Annotation layout" and "Annotation types" groups, respectively, the location of the conserved amino acid can be visualized (see figure 8.6).
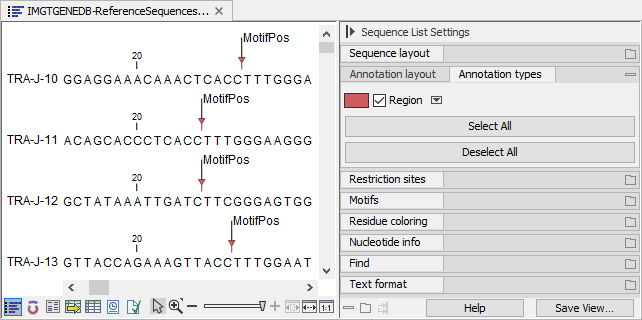
Figure 8.6: Visualizing the location of the conserved amino acid.
For the IMGT format, the sequence list also contains the accession number(s) and species. These can be seen in the table view (see figure 8.7).
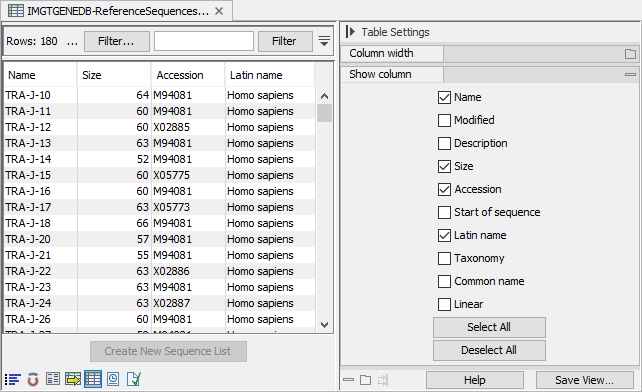
Figure 8.7: Table view of imported sequence list showing the name, species and accession number when imported using the IMGT format.