Identify Mispriming Events
Primers with high similarity to multiple genomic regions have the potential to be involved in mispriming, where reads are amplified from a region of the genome other than the intended target region. Reads resulting from such mispriming events are fused constructs: the primer part and the read part represent different regions of the genome.
A fraction of the population of a given primer may be involved in mispriming events, resulting in low frequency variants. How large this fraction is depends on the binding affinity and specificity of the primer, and the conditions that the lab work was performed under. Reads originating from mispriming events should be identified and removed from mappings to avoid calling false positive variants. If reads from mispriming events map to the region they originate from, and non-target regions of interest are known, then the primer can be unaligned, rather than removing the whole read.
Identify Mispriming Events generates a list of potential mispriming events for a set of panel primers and a specified reference genome. This list of mispriming events can then be supplied to Trim Primers of Mapped Reads to remove reads likely to represent a mispriming event, or to unalign primer parts of such reads, as relevant. This should precede variant detection, so as to minimize false positive variant calls due to artifacts generated from mispriming events.
Remove reads or unalign primer regions?
Trim Primers of Mapped Reads can handle misprimed reads when provided with a track of predicted mispriming events. Reads are either removed completely from the read mapping or having their primer region unaligned. This is done automatically during primer trimming to avoid calling false positiv variants, and the action needed depends on where reads resulting from a mispriming event are mapped:
- To the original, intended target region Reads amplified from a non-target region may still map to the original, intended target region if it has sufficient similarity to that region.
- Symptom: No mismatches in the primer region in the mapping. Mismatches in the non-primer region if the sequence downstream of the intended primer site and downstream of the mispriming site differ.
- Action to take: Such reads should be removed from the read mapping before calling variants, since the reads are mapped to a different region than the one they are amplified from.
- To the non-target region it represents The read maps best to the non-target region it was generated from.
- Symptom: Mismatches in the primer region of the read (unless the mispriming event has 100% identity). No mismatches in the non-primer region (besides mismatches due to true variants).
- Action to take: Unalign the primer part of such reads in regions relevant for calling variants. Even if the primer part is 100% identical to the non-target region sequence, unaligning the primer part of these reads is important, as this allows the correct frequencies of variants in that region to be determined. If unaligning primer regions in this circumstance is desired, a track containing the regions where variant calling is of interest must be provided when launching this tool.
See figure 6.83 for an example.
A given primer can be involved in mispriming events leading to the amplification of reads that map to the original target region and to the region they were amplified from.
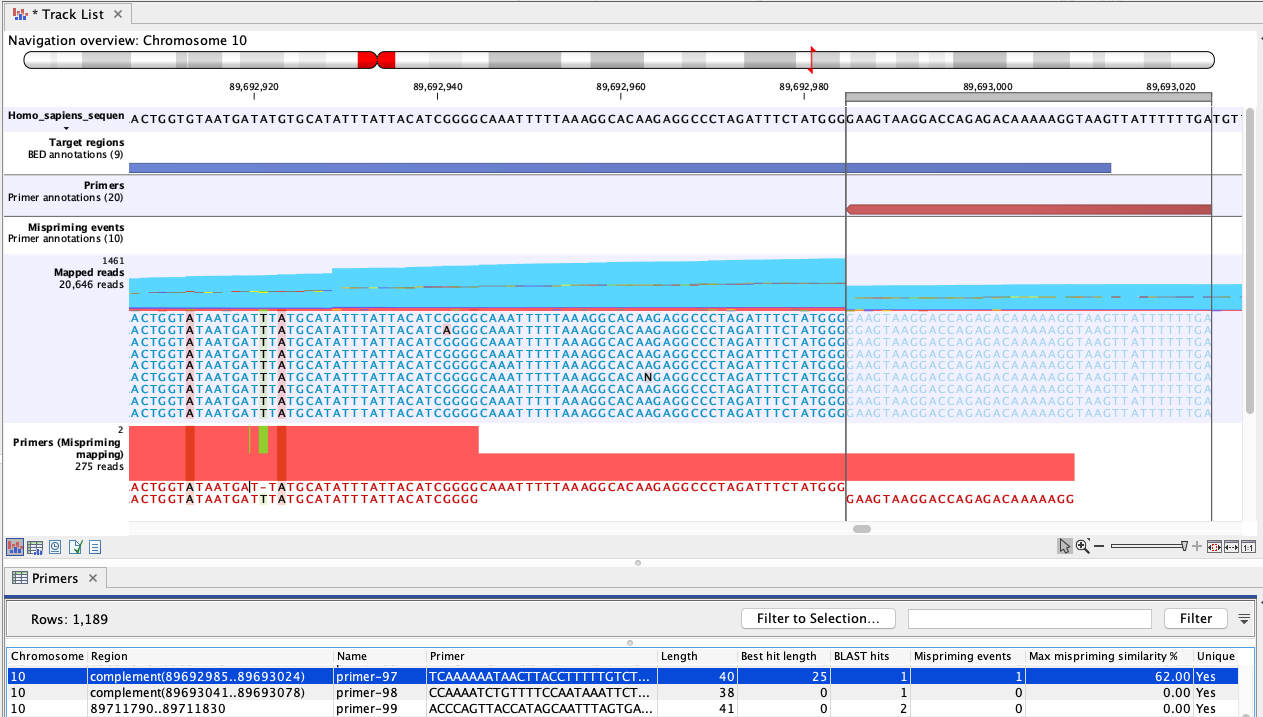
Figure 6.82: An example of mispriming, where the reads map to the original intended target region. The two A variants and the single T variant, occuring in a non-primer part of the mapped reads, are consequences of mispriming. The reads supporting these variants should be removed from the mapping before variant detection is carried out. The reverse paired end reads (light blue) shown in the "Mapped reads" track were amplified from a mispriming binding site at chromosome 9 (not shown). While the primer had only 62% similarity with that site, the 3' primer end aligned perfectly, allowing it to anneal and for reads to be generated. Most of these reads mapped to the original target region, shown here, due to the low similarity of the primer region and high overall similarity with the intended target region.
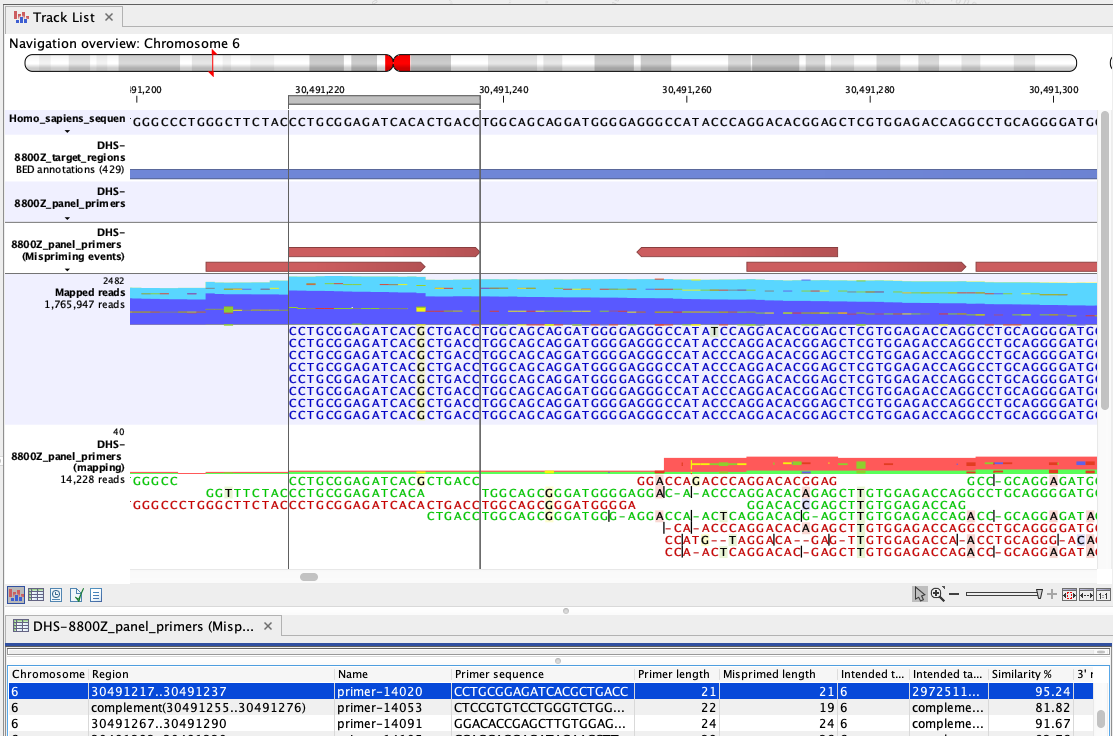
Figure 6.83: An example of mispriming, where the reads map to the non-target region it represents. The A to G variant, found in the primer part of these forward, paired end reads (dark blue), is a consequence of mispriming. This primer was designed for a different region, but had 95.24% similarity with the region shown. Thus some copies of the primer annealed to this region and generate reads with a single mismatch, as shown in the "Mapped reads" track. The primer part of these reads should be unaligned, but the remaining part of the read can still be used for variant calling since it reflects the DNA fragments of the same genomic region.
How the Identify Mispriming Events tool works
Identify Mispriming Events takes this approach:
- A BLAST search (blastn-short) is run using the primers as query sequences to search against a BLAST database of the relevant reference genome.
- The BLAST hits returned are filtered. For each primer, hits are kept if that sequence has a high enough similarity to the intended target region and few mismatches at the 3' end.
- The remaining BLAST hits for each primer are checked for their potential to cause mispriming artifacts of the two types mentioned above.
- The sequence downstream of the intended target binding site is aligned to the sequence downstream of the mispriming site. If this pairwise alignment has a similarity fraction of at least 0.8, the BLAST hit is considered to be a mispriming event. The length of the sequence used of alignment can be changed using the parameter Amplicon length (bp).
- If a target region track is provided as input, and the mispriming region overlap a target region, the BLAST hit is considered a mispriming event.
Running the Identify Mispriming Events tool
To launch Identify Mispriming Events, go to:
Toolbox | QIAseq Panel Expert Tools (![]() ) | QIAseq DNA Panel Expert Tools (
) | QIAseq DNA Panel Expert Tools (![]() ) | Identify Mispriming Events (
) | Identify Mispriming Events (![]() )
)
In the first dialog, select a primer track (![]() ) as input.
) as input.
Figure 6.84: Select a primer track.
Settings related to the reference data are configured in the next wizard step (figure 6.85).
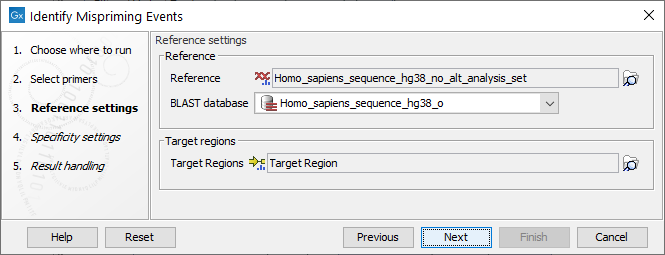
Figure 6.85: Reference data settings for the Identify Mispriming Events tool.
- Reference: A reference sequence track (
 ) compatible with the selected primer track. Primer sequences are extracted from this reference. If a track is supplied in the "Target region track" field below, those regions are also extracted.
) compatible with the selected primer track. Primer sequences are extracted from this reference. If a track is supplied in the "Target region track" field below, those regions are also extracted.
- BLAST database: A BLAST database of the selected reference genome.
- Target regions: An annotation track (
 ) containing regions known to be of interest for variant calling. This track is required to obtain the type of mispriming events with mismatches in the primer region of the mapped read, where primer regions should be unaligned before variant calling.
) containing regions known to be of interest for variant calling. This track is required to obtain the type of mispriming events with mismatches in the primer region of the mapped read, where primer regions should be unaligned before variant calling.
Tip: Use Create BLAST Database if you do not already have a BLAST database of your reference genome (see
https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Create_local_BLAST_databases.html
). This tool takes a sequence list (![]() ) as input. If your reference genome is in a sequence track (
) as input. If your reference genome is in a sequence track (![]() ), use Convert from Tracks to convert it to a sequence list (
), use Convert from Tracks to convert it to a sequence list (![]() ) before running Create BLAST Database.
) before running Create BLAST Database.
Specificity settings are configured in the next wizard step (figure 6.86).
Figure 6.86: The specificity settings of the Identify Mispriming Events tool. The default settings are a good starting point, but BLAST settings and/or Mispriming events filters can be adjusted to make the settings more relaxed or stringent.
- BLAST word size: An exact match of at least this length between the primer and reference genome is required for BLAST to initiate an extension that might lead to a reported hit. This value should be set based on the shortest primer in the primer set, and the word size should never be longer than half the length of the shortest primer. Increasing this value increases specificity, but too high a value could result in potential mispriming sites not being reported.
- BLAST expect value: Lower expect values are more stringent, leading to fewer chance matches being reported. However, virtually identical short alignments have relatively high values because of the way expect values are calculated. Raising this value can lead to more potential mispriming sites being identified, but can also increase the running time of the tool.
- Maximum number of BLAST hits per chromosome: The maximum number of BLAST hits to report per chromosome. Limiting the number of hits to return can decrease the time the running time of the tool and circumvent potential out-of-memory issues.
- Minimum similarity %: The minimum percentage similarity between a primer sequence and non-target regions in the reference genome for that region to be retained in the list of potential mispriming event sites.
- Maximum mismatches in 3' end: The number of mismatches allowed between a primer and a non-target region of the reference genome, counting from the 3' end of the primer. If this value is exceeded, the region is not retained in the list of potential mispriming event sites.
- Amplicon length (bp): The length of sequence from downstream of the designed primer site and from downstream of the mispriming site that should be used to test if reads amplified from the mispriming site has the potential to map to the original, intended target region can cause false positive variant calls. If the pairwise alignment of these two regions has a similarity fraction of at least 0.8, the region is reported as a mispriming event.
Output from Identify Mispriming Events
Four outputs are produced from the Identify Mispriming Events tool:
- Mispriming events: An annotation track that can be used for Trim Primers of Mapped Reads. The track has a row for each mispriming event.
- Primers: An annotation track of primers annotated with different mispriming statistics.
- Misprimed reads track: A read mapping of reads representing mispriming events.
- Report: A report that summarizes the mispriming events identified by the tool.
Mispriming events
The mispriming events track includes the following annotations:
- Primer sequence: The sequence of the primer.
- Primer length: The length of the primer.
- Misprimed length: The length of the mispriming site where the primer sequence aligns.
- Intended target chromosome: The chromosome that the primer was designed for.
- Intended target region: The region that the primer was designed for.
- Similarity %: Similarity percentage between the primer sequence and the sequence of the mispriming site.
- 3' mismatches: The number of nucleotide mismatches before the first match in the 3' primer end.
- Primer part mismatch type: Yes if the mispriming event potentially causes false positives in the primer part of the mapped read, otherwise No.
- Non-primer part mismatch type: Yes if the mispriming event potentially causes false positives in the non-primer part of the mapped read, otherwise No. Only evaluated if a target region track is provided.
Primers
The primer track includes the following annotations:
- Length: The length of the primer.
- Best hit length: The length of the BLAST hit with the highest similarity percentage.
- BALST hits: Number of filtered BLAST hits for this primer.
- Mispriming events: Number of mispriming events for this primer.
- Mispriming events > 80%: Number of mispriming events with a similarity percentage of at least 80% for this primer.
- Mispriming events > 90%: Number of mispriming events with a similarity percentage of at least 90% for this primer.
- Mispriming events with non-primer part mismatches: Number of mispriming events, originating from this primer, that potentially cause mismatches in the non-primer part of the mapped read.
- Mispriming events with primer part mismatches: Number of mispriming events, originating from this primer, that potentially causes mismatches in the primer part of the mapped read. Only evaluated if a target region track is provided.
- Max mispriming similarity %: Maximum similarity percentage among the identified mispriming events for this primer.
- Unique primer: Yes if the primer is unique in the reference genome, No if the primer has a 100% similarity match to another genomic region.
Misprimed reads track
Each mispriming event is represented by two reads in the read mapping: A read with the sequence of the primer aligned to the mispriming site, and a read with the sequence of the mispriming site aligned to the primer design region. Primers with multiple mispriming events will have a read for each mispriming event aligned at the primer design region.
For mispriming events that potentially cause false positives in the non-primer part of the read, two additional reads are included in the read mapping: A read with the downstream sequence of the primer aligned to the downstream of the mispriming site, and a read with the downstream sequence of the mispriming site aligned to the downstream of the primer design region. The mismatches in these reads show the potential false positive variants that can arise from mispriming.
Report
The mispriming event report includes the following information:
- Summary: A summary table showing the number of input primers and input target regions, as well as how many primers that have mispriming events and the types of potential false positives.
- Primers with potential mispriming: The section provides information about the primers for which one or more mispriming events have been found. The number of BLAST hits and the number of mispriming events for each primer are shown as distribution plots, as well as the maximum mispriming similarity percentage for these primers.
- Mispriming events: Different statistics about the BLAST hits and mispriming events identified by the tool.