Remove and Annotate with Unique Molecular Index
During library preparation of the samples it is possible to add single or duplex UMI sequences to the reads, which are used towards correcting for sequencing errors and to help improve performance. Addition of UMI is often accompanied by a common sequence prefix that is also added before amplification and which can be very helpful when locating the exact UMI sequence. While the UMI is essential in identifying reads that originate from the same fragment, retaining it as such on the sequenced reads would hinder the subsequent read mapping efficiency and accuracy. Therefore, the Remove and Annotate with Unique Molecular Index tool removes the UMI and the common sequence prefix from the reads, while annotating each read with the UMI to retain the fragment identity as annotation.
The tool can be found in the Toolbox here:
Tools | QIAseq Panel Expert Tools (![]() ) | QIAseq DNA Panel Expert Tools (
) | QIAseq DNA Panel Expert Tools (![]() ) | Remove and Annotate with Unique Molecular Index (
) | Remove and Annotate with Unique Molecular Index (![]() )
)
In the first dialog, select sequence list(s) (![]() ) containing the reads.
) containing the reads.
In the Settings dialog (figure 6.65), the following options are available:
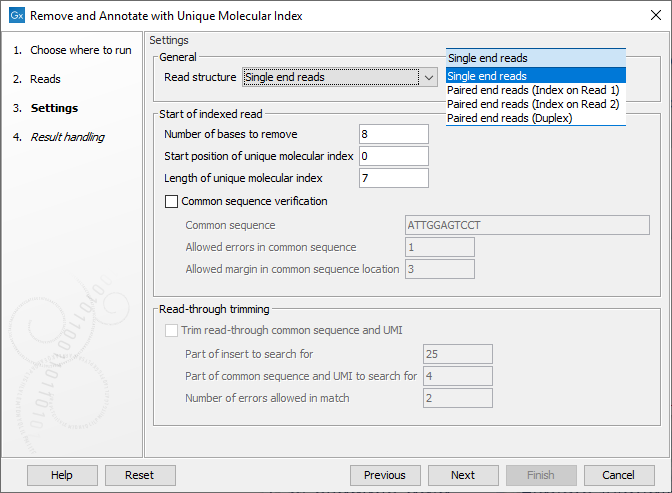
- Read structure Specify the read type and, for paired data, which read the UMI is present on or if there are duplex UMI. The duplex option assumes the TruSight Oncology, Illumina (TSO) UMI protocol was used.
- Number of bases to remove The length of the UMI, or duplex UMI, plus the length of the common sequence. The default value is 8 for duplex UMI and 23 for other cases.
- Start position of Unique Molecular Index The initial position of the UMI on the read. The default is 0, indicating that the UMI is at right at the start of the reads.
- Length of Unique Molecular Index The length of the UMI. The default value is 7 for duplex UMI and 12 for other cases.
- Common sequence verification When enabled, a common sequence to be searched for, next to the UMI on each read, can be specified. By default, this option is unchecked, and the tool will output the same number of reads as were present in the input, but with UMI and common sequences trimmed away. When checked, only reads containing the specified common sequence, within the given error margins, will be retained. The following fields must be filled in when using this option:
- Common sequence The common sequence to be searched for.
- Allowed errors in common sequence The number of insertion/deletion/mismatches allowed between the common sequence defined, and the read sequence, for that read to be retained.
- Allowed margin in common sequence location The number of base positions that the actual location of the common sequence can differ from its intended location.
- Trim read through common sequence and UMI: If this option is enabled, then for each read pair, first a sequence is extracted from the indexed read consisting of a part of the insert sequence and a part of the adjacent common sequence and UMI. Then, the reverse complement of this sequence is used to search the non-indexed read of a read pair, and if a match is found, the non-indexed read will be trimmed at the boundary between the insert and the common sequence.
- Part of insert to search for Number of nucleotides from the sample sequence insert used to identify read-through. Increase this value to get more specific matches, decrease it if the indexed reads are very short, or to improve speed.
- Part of common sequence and UMI to search for Number of nucleotides from the common sequence and UMI used to identify read-through. Increase this value to get more specific matches and avoid truncation at repetitive instances, decrease it to trim off shorter partial occurrences of common sequence and UMI.
- Number of errors allowed in match Number of insertion, deletion, or mismatch errors allowed when looking for read-through sequences.
A report can be generated that contains information about the number of reads processed, and the number and fraction of reads found to have UMIs. It also includes a plot of the nucleotide distribution per position of the UMI barcode.