Data QC and Clean Host DNA
The Data QC and Clean Host DNA workflow performs trimming of reads, creates a QC report and cleans the dataset from host DNA, leaving back only the reads that do not match the host genome.
To run the Data QC and Clean Host DNA workflow, go to Metagenomics (![]() ) | Taxonomic Analysis (
) | Taxonomic Analysis (![]() ) | Workflows | Data QC and Clean Host DNA.
) | Workflows | Data QC and Clean Host DNA.
You can select only one read file to analyze (figure 6.15). Alternatively, multiple files can be run using the batch mode.
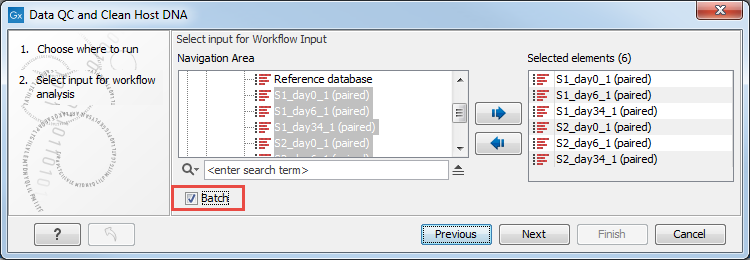
Figure 6.15: Select the reads.
In the "Trim Sequences" dialog, you can specify a trim adapter list and set up parameters if you would like to trim your sequences from adapters (figure 6.16).
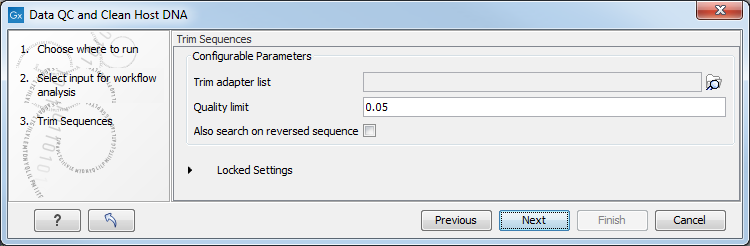
Figure 6.16: You can choose to trim adapter sequences from your sequencing reads.
The parameters that can be set are:
- Quality limit: defines the minimal value of the Phred score for which bases will not be trimmed.
- Also search on reversed sequence: the adapter sequences will also be searched on reverse sequences.
In the "Taxonomic Profiling" dialog, select the reference database you will use to map the reads (figure 6.17). It is possible to "Filter host reads". You must then specify the host genome (in the case of human microbiota, the Homo sapiens hg38 for example).
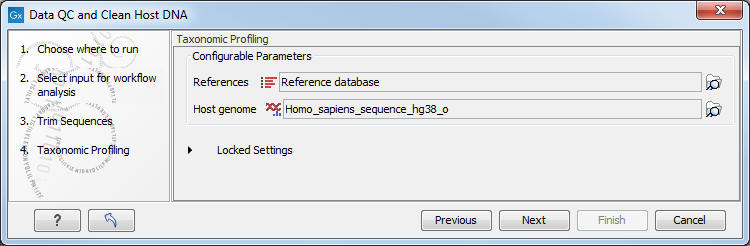
Figure 6.17: Select the reference databaes, and potentially choose to filter against an host genome to remove possible contamination.
The workflow will output a sequence list with reads cleaned from host DNA. In addition, it generates three reports: a trimming report, a graphical QC report and a supplementary QC report. All of these should be inspected in order to determine whether the quality of the sequencing reads and the trimming are acceptable. For a detailed description of the QC reports and indication on how to interpret the different values, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=QC_Sequencing_Reads.html.
For the trimming report, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Trim_output.html.