Running Detect and Refine Fusion Genes
To run the tool, go to:
Tools | RNA-Seq and Small RNA Analysis (![]() )| RNA-Seq Tools (
)| RNA-Seq Tools (![]() ) | Detect and Refine Fusion Genes (
) | Detect and Refine Fusion Genes (![]() )
)
The tool takes as input one or more sequence lists (![]() ) containing reads.
) containing reads.
Selecting references
The following options can be configured in the References dialog (figure 33.56):
- RNA-Seq reads track. A reads track (
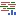 ) produced by RNA-Seq Analysis, where the same input reads (
) produced by RNA-Seq Analysis, where the same input reads ( ) were used.
) were used.
- Reference sequence. A sequence track (
 ) with the wildtype (WT) reference genome.
) with the wildtype (WT) reference genome.
- Gene track. An annotation track (
 ) with the WT gene annotations.
) with the WT gene annotations.
- mRNA track. An annotation track () with the WT transcript annotations.
The same tracks as used for RNA-Seq Analysis should be provided.
- CDS track. An optional annotation track () with WT coding sequence annotations.
- Primer track. An optional annotation track () with the WT primer regions.
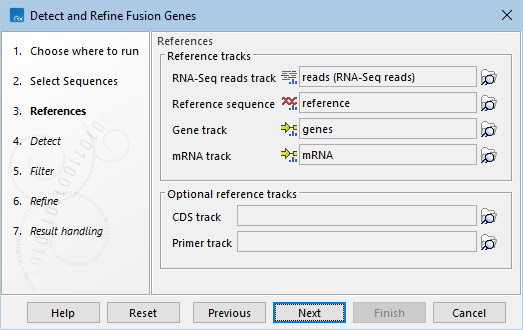
Figure 33.56: Reference tracks for Detect and Refine Fusion Genes.
Detecting fusions
Potential fusions are detected by:
- Identifying unaligned ends in gene regions from the input 'RNA-Seq reads track'.
- Mapping the unaligned ends to the WT reference genome using 'RNA-Seq Analysis' with default options.
- Determining potential fusions using the unaligned ends that map within gene regions.
- A potential fusion consists of a 5' and 3' gene.
- A fusion between a 5' and 3' gene is defined by a pair of breakpoints: the 5' breakpoint is located at the beginning of the unaligned end in the input 'RNA-Seq reads track', while the 3' breakpoint is at the beginning of the unaligned end mapped to the WT reference genome (figure 33.55).
- For a given 5' and 3' gene pair, multiple fusions with distinct breakpoint pairs may be identified.
The following options can be configured in the Detect dialog (figure 33.57):
- Minimum length of unaligned ends. Only unaligned ends longer than this are used to detect fusions.
- Maximum distance to known exon boundary. Potential fusions are built using unaligned ends that:
- Are located in the input 'RNA-Seq reads track' within this distance of a known exon boundary.
- Map to the WT reference genome within this distance of another known exon boundary.
The fusion breakpoints are placed at the respective exon boundaries.
The signal for fusions can be increased by using larger maximum distances, as unaligned ends farther from known exon boundaries are then also considered. However, this also decreases the resolution, as transcripts with exon boundaries within this distance of each other would not be differentiated. This can reduce the number of mapped reads in the refinement step.
- Detect exon skippings. When checked, fusions involving the same gene, corresponding to transcripts that have missing exons compared to the WT transcript annotation, are detected.
- Detect novel exon boundaries. When checked, fusions with one breakpoint located farther than 'Maximum distance to known exon boundary' from an exon boundary are also detected.
- Detect novel exon boundaries in both genes. When checked, fusions with both breakpoints farther than 'Maximum distance to known exon boundary' from an exon boundary are also detected.
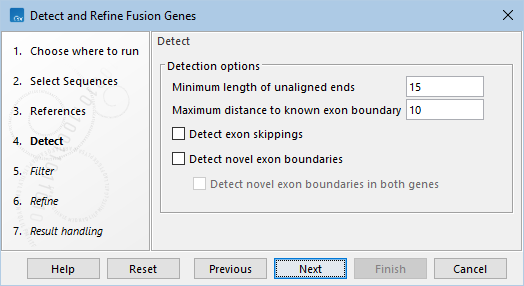
Figure 33.57: Default options for detecting fusion genes.
Filtering fusions
Candidate fusions are identified among the detected potential fusions by:
- Optionally filtering using lists of genes and/or fusions.
- Applying a series of filters:
- The best scoring fusions, based on p-value and Z-score, are selected.
- The number of breakpoint pairs for one gene pair is capped at 20.
- Multiple breakpoint pairs for the same gene pair are treated as one candidate fusion.
The following options can be configured in the Filter dialog (figure 33.58):
- Gene filter action. How detected fusions should be filtered based on the 5' and 3' genes. The following filter actions are available:
- None. Detected fusions are not filtered.
- Exclude. Detected fusions with a 5' or 3' gene in the provided track or list are removed. This can be useful for removing unwanted fusions.
- Include. Only detected fusions with a 5' or 3' gene in the provided track or list are kept. This can be useful for restricting fusion detection to only genes of interest.
The genes used for filtering can be provided using at least one of the following options:
- Genes for filtering (tracks). A gene track () that is a subset of the reference gene track. The gene IDs are used for filtering. Can be left empty or multiple tracks can be provided.
Several tracks suitable for excluding genes are available via the Reference Data Manager, see Exclude lists for details.
- Genes for filtering (names). A list of case-sensitive gene IDs or names separated by comma, semicolon, or any white-space character. Can be left empty.
Additionally, the following can be configured:
- Require both genes. When checked, both 5' and 3' genes must be in the provided track or list in order for the fusion to be filtered.
- Fusion filter action. How detected fusions should be filtered. The following
filter actions are available:
- None. Detected fusions are not filtered.
- Exclude. Detected fusions in the provided table or list are removed. This can be useful for removing fusions that are frequently detected as false positives.
- Include. Only detected fusions in the provided table or list are kept. This can be useful for restricting fusion detection to only fusions of interest.
The fusions used for filtering can be provided using at least one of the following options:
- Fusions for filtering (tables). A table (
 ) with case-sensitive gene IDs or names. Can be left empty or multiple tables can be provided. The following formats are supported:
) with case-sensitive gene IDs or names. Can be left empty or multiple tables can be provided. The following formats are supported:
- The first two columns contain one gene each.
- The first column has a header with the word 'fusion' (case-insensitive) and contains gene pairs in the format 'gene1-gene2'.
- The first column has a header with the word 'genes' (case-insensitive) and contains a group of genes in the format 'gene1 gene2 gene3'. Each group results in all possible gene combinations, such as 'gene1-gene2', 'gene1-gene3', and 'gene2-gene3'.
All other columns are ignored.
See Standard import for details on how to import tables.
Several tables suitable for excluding fusions are available via the Reference Data Manager, see Exclude lists for details.
- Fusions for filtering (names). A list of case-sensitive gene IDs or names in the format 'gene1-gene2' separated by comma, semicolon, or any white-space character. Can be left empty.
If the IDs or names of genes in the fusions used for filtering contain a '-', the 'Fusions for filtering (tables)' using the second format ('fusion' header) and 'Fusions for filtering (names)' cannot be used. We recommend choosing one of the alternative options instead.
The fusions used for filtering are non-directional: a gene pair 'gene1-gene2' applies to fusions where gene1 is the 5' gene and gene2 is the 3' gene, as well as fusions where gene2 is the 5' gene and gene1 is the 3' gene.
- Assumed error rate. The probability of the binomial distribution used to calculate p-values. For example, an assumed error rate of 0.001 indicates that, on average, 1 in 1000 reads covering a breakpoint supports the fusion by chance.
The default value is deliberately conservative, which may result in low-frequency fusions not being selected as candidate fusions. By decreasing the value, more candidate fusions can be identified.
- Maximum number of fusions. The maximum number of candidate fusions.
- Minimum unaligned end count. The minimum number of unaligned ends that must support a candidate fusion.
- Promiscuity threshold. The maximum number of candidate fusions for a given gene.
- Include all fusions in the WT track output. If checked, detected potential fusions that were not selected as candidate fusions are included in the 'Fusion genes, WT' track output. See Filtered fusions for details.
Figure 33.58: Default options for filtering fusion genes.
Refining fusions
Once candidate fusions have been identified, their support is further refined by:
- Creating an artificial fusion genome with one chromosome for each candidate fusion. Corresponding transcripts for all detected breakpoints are created, along with tracks for genes, and, if available, CDS and primers.
- Mapping the input reads simultaneously to the WT and artificial fusion genomes, allowing them to be assigned to the genome (WT or artificial fusion) where they map most accurately.
- Counting the number of reads supporting the fusion vs the WT genes, and recalculating p-values and Z-scores.
- Evaluating the support for candidate fusions for each identified breakpoint pair, and assigning a PASS status to breakpoint pairs if they are found to be adequately supported.
The following options can be configured in the Refine dialog (figure 33.59):
- Mapping options. Options for mapping the input reads to the WT reference genome and artificial fusion genome. See Mapping settings for details.
- Minimum number of supporting reads. The minimum number of reads required for a PASS status.
- Maximum p-value. The maximum allowed p-value for a PASS status.
- Minimum Z-score. The minimum required Z-score for a PASS status.
- Breakpoint distance. The minimum distance from the ends of a read to the breakpoint required for supporting reads. It corresponds to the minimum number of bases that a read must span on each side of the breakpoint.
- Report only significant breakpoints. When checked, only breakpoint pairs with a PASS status are added to the report.
Figure 33.59: Default options for refining fusion genes.
The default values for 'Maximum p-value' and 'Minimum Z-score' are deliberately conservative, which may result in low-frequency fusions not being assigned a PASS status. By increasing 'Maximum p-value' and/or decreasing 'Minimum Z-score', more fusions can be identified.