Create K-medoids Clustering for RNA-Seq
In a k-medoids clustering, features are clustered into k separate clusters. The procedure seeks to assign features to clusters such that distances between features of the same cluster are small, while distances between clusters are large.
The output of the tool is a Clustering Collection (![]() ). The clusters in the Clustering Collection can be viewed together as a Sankey plot (
). The clusters in the Clustering Collection can be viewed together as a Sankey plot (![]() ) or individually as graphs (
) or individually as graphs (![]() ).
).
To perform a k-medoids clustering:
Tools | RNA-Seq and Small RNA Analysis (![]() )| Expression Plots (
)| Expression Plots (![]() ) | Create K-medoids Clustering for RNA-Seq (
) | Create K-medoids Clustering for RNA-Seq (![]() )
)
Select at least two expression tracks (![]() ), or miRNA expression tables (
), or miRNA expression tables (![]() )/ (
)/ (![]() ).
).
Click Next to display a dialog as shown in figure 33.77.
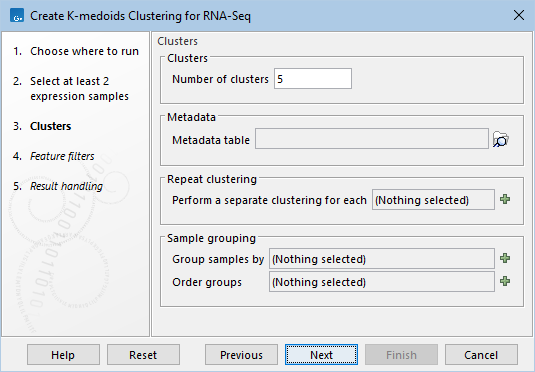
Figure 33.77: Parameters for k-medoids clustering.
The parameters are:
- Number of clusters. The maximum number of clusters to cluster features into: the final number of clusters will be smaller than this if there are fewer features than clusters.
- Metadata table (Optional) The metadata table describing the factors for the selected inputs.
- Perform a separate clustering for each (Optional) one of the factors from the metadata table. A separate k-medoids clustering is performed for each group in this factor. The clusters for each group form separate columns in the Sankey plot. This is useful when looking for genes whose expression pattern changes in a certain way between groups. The groups could, for example, represent different treatments.
- Group samples by (Optional) One of the factors from the metadata table. The distances between samples for a feature are calculated using the group means. If this is left blank, then distances will be calculated using all the individual values of the samples.
- Order groups (Optional) For the chosen Group samples by, specify the order of the groups. The ordering controls the x-axis of the expression graphs. This is useful when the data has a natural ordering, such as a time series. If only some groups are ordered here, then these will come first, and the remaining groups will be added at the end.
- No filtering. All features are kept.
- Fixed number of features. A specified number of features are kept.
- Number of features. This number of features with the highest index of dispersion (variance to mean ratio) are kept. Raw count values (not normalized) are used to calculate this index.
- Minimum counts in at least one sample. Only features with at least this raw count value in one sample are kept.
- Filter features by statistics. Features that are differentially expressed according to the specified thresholds are kept.
- Statistical comparison. A Statistical Comparison Track (
 ), see Output of the Differential Expression tools.
), see Output of the Differential Expression tools.
- Minimum absolute fold change. Only features with at least this absolute fold change are kept.
- Threshold. Only features with at most this p-value are kept. The type of the p-value can be set to P-value, FDR p-value, or Bonferroni.
- Statistical comparison. A Statistical Comparison Track (
- Specify features. A set of specified features are kept.
- Feature track. An Annotation Track (
 ). All features in this track are kept.
). All features in this track are kept.
- Feature names. Features with names in the list of case-sensitive names are kept. Any white-space characters, comma, and semicolon are accepted as separators.
- Feature track. An Annotation Track (
We only recommend using Fixed number of features for exploratory analysis. This is because, while the chosen features have the most variable expression among all the samples, the variation may not be of interest: for example, maybe there is a large variability across different time points in a time series, but this is the same in both treatment and control groups.
Subsections