Structural Variant Caller
The Structural Variant Caller identifies structural variants in read mappings based on evidence from unaligned read ends and coverage information. It builds on the same ideas around unaligned end read signatures as the existing InDels and Structural Variants tool, but to a larger extent relies on statistical reasoning and more refined components for consensus generation, mapping and alignment of the unaligned end sequences.
The tool,
- detects deletions, insertions (including tandem duplications), and inversions.
- is applicable to read mappings of Targeted, Exome, and WGS (Whole Genome Sequencing) NGS resequencing data.
- is developed for short read technologies (such as Illumina reads).
- detects germline as well as somatic variants.
The tool has the following limitations:
- Inter-chromosomal rearrangements are not supported.
- In read mappings of RNA-Seq data each part of a spliced read is treated independently.
The tool processes each chromosome in a genome individually, through several steps:
Breakpoint estimation: The tool looks for unaligned read ends at each chromosome position. Consensus sequences are constructed for the unaligned ends and aligned regions across the reads at a breakpoint (one consensus sequence for the unaligned end and one for the aligned region). The consensus sequence is based on a majority count of k-mers for the unaligned end, while the nucleotide count in each column is used for the aligned region. Breakpoints are labeled either as a 'left' or 'right' breakpoint. This labeling is from the perspective of a deletion, where a left breakpoint is on the left side of a deletion (which means there is a right unaligned end) and a right breakpoint is vice-versa on the right side of the deletion. For WGS applications, the tool makes a probabilistic assessment of how likely the breakpoint is to support a structural variant based on the coverage, the unaligned end read count, and the specified ploidy of the sample.
Coverage and complexity estimation (WGS applications only): each chromosome is divided into bins. The tool then calculates the coverage and the complexity of the reference region in each bin. The complexity is calculated using the Lempel-Ziv complexity measure and is used to avoid calling structural variants in low-complexity regions, while the coverage information, is stored and, together with breakpoint information, used to find potential copy number variations (see below).
Resolving structural variants: after breakpoints have been established, different combinations of left and right breakpoints are paired together. For each pair, the unaligned and aligned consensus sequences from one breakpoint are aligned to the other breakpoint. The alignment scores from each possible pairing are then stored in a matrix, and a dynamic programming algorithm is used to identify which breakpoints to pair together. Breakpoints that were not matched in this step are then each used as a single breakpoint to search for additional structural variants, either in terms of (1) smaller insertions or deletions inferred from self-mapping evidence (where the unaligned consensus itself maps back to nearby its own location), or (2) as supporting evidence for CNV losses or gains inferred from the coverage analysis (see below).
Copy number variation (WGS applications only): the counts in each bin along the chromosome are used to determine if there is a statistically significant difference when compared with a normal distribution, where the normal distribution is modeled on the basis of the mean and standard deviation for the counts across all the bins (this part of the algorithm is based on [Yoon et al., 2009]). When there is a consecutive number of statistically significant bins, they are combined using Fisher's method to calculate a total significance value, which is then used to determine if there is a CNV. Each CNV is then used with any nearby breakpoints to determine if there is a deletion or duplication present.
Running the Structural Variant Caller tool
To run the Structural Variant Caller tool, go to:
Toolbox | Resequencing Analysis (![]() ) | Variant Detection (
) | Variant Detection (![]() ) | Structural Variant Caller (
) | Structural Variant Caller (![]() )
)
Once the tool wizard has opened (figure 24.18), choose the read mapping you would like to analyze. The Structural Variant Caller tool accepts read mappings as either reads tracks or stand-alone read mappings.
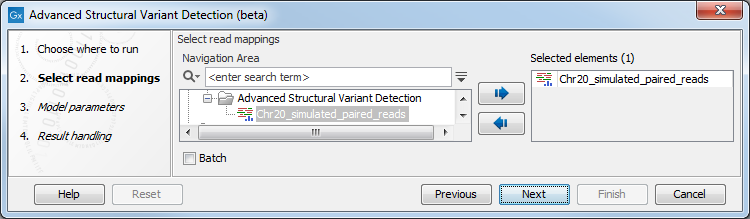
Figure 24.18: Select one or several reads tracks or stand-alone read mappings.
In the next wizard step, specify the ploidy and application for the sample you are analyzing (figure 24.19). You can also specify to ignore broken pair reads. Ignoring broken pairs will typically reduce the computational time of the analysis. It may have a negative impact on sensitivity, but may also improve precision, depending on the source of the broken pair reads.
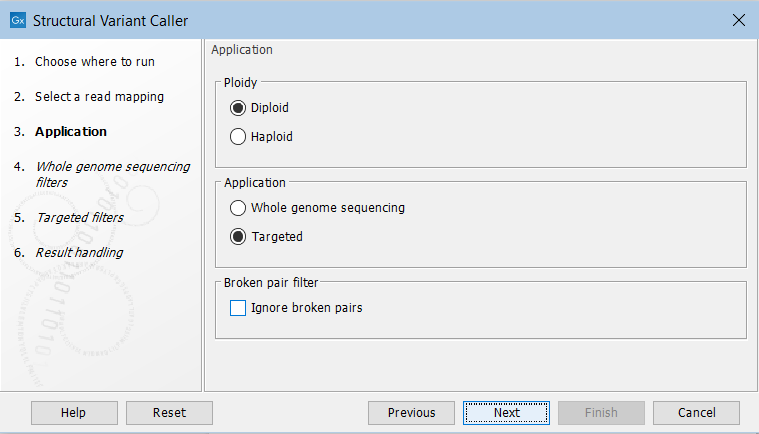
Figure 24.19: Set the application parameters for the tool and specify if broken pair reads should be ignored.
- Ploidy Specifies the ploidy of the sample. The value determines the maximum number of overlapping structural variants that can be detected, and, when Whole Genome Sequencing is specified as application, is also used for calculating breakpoint probabilities. Diploid should be chosen unless the data is from a haploid organism.
- Application Choose "Targeted" if running on a read mapping of targeted or whole exome sequencing data and otherwise choose "Whole Genome Sequencing". When Targeted is chosen the coverage and complexity analysis is not applied (it relies on model assumptions that are only appropriate for WGS applications) .
In the next steps you are asked to specify filter settings. The settings depend on whether you have specified the whole genome sequencing or the targeted application. The filter settings for the whole genome sequencing application (figure 24.20) are:
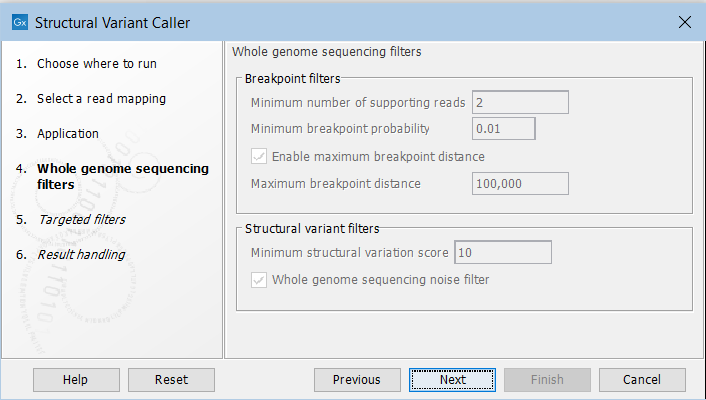
Figure 24.20: Set filters for the whole genome sequencing applications.
- Minimum number of supporting reads Minimum number of reads with unaligned ends required for a breakpoint to be considered.
- Minimum breakpoint probability Minimum required probability of a breakpoint to be considered (based on a statistical model and only applicable to Whole Genome Sequencing data).
- Maximum breakpoint distance If enabled, structural variants cannot be detected when a pair of breakpoints are further apart than this value. As most of the detected structural variants are found using breakpoint pairs, the maximum length of detected deletions, tandem repeats, and inversions will therefore typically be limited by this distance (note that re-alignment of a detected structural may occur, in which case the variant can extend beyond the breakpoint positions). Higher breakpoint distances will increase processing time, but will allow for the detection of longer deletions, tandem repeats, and inversions.
- Minimum structural variation score Measure of the overall evidence supporting the structural variant detected. The value is based on the alignment scores of the unaligned ends or, in case of shorter indels, the length of the variation. This value may be increased to reduce the number of structural variants called.
- Whole genome noise sequencing filter In the case WGS data, a number of different filters are used to remove unlikely structural variants. The filters are based on information such as the coverage, length and complexity of unaligned ends, and also variant and breakpoint probabilities.
For the targeted application the filters are (figure 24.21):
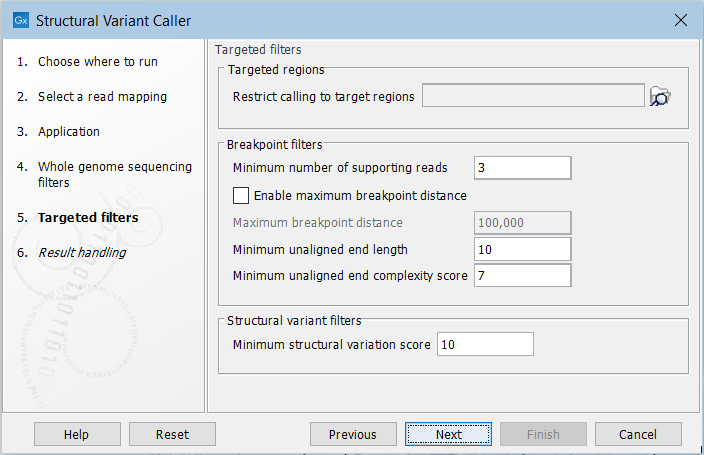
Figure 24.21: Set filters for the targeted sequencing applications.
- Targeted regions Allows you to specify an annotation track, to which the analysis will be restricted. When this is done, only breakpoints located within the specified regions (with a buffer of 15bp) will be considered and structural variant calls will be limited to those that are inferred from these breakpoints. This will typically decrease computational time, but may also cause variants with evidence located outside the specified regions to go undetected.
- Minimum number of supporting reads Minimum number of reads with unaligned ends required for a breakpoint to be considered.
- Maximum breakpoint distance If enabled, structural variants cannot be detected when a pair of breakpoints are further apart than this value. As most of the detected structural variants are found using breakpoint pairs, the maximum length of detected deletions, tandem repeats, and inversions will therefore typically be limited by this distance (note that re-alignment of a detected structural may occur, in which case the variant can extend beyond the breakpoint positions). Higher breakpoint distances will increase processing time and may make it more difficult to find smaller variants, but will allow for longer deletions, tandem repeats, and inversions to be detected.
- Minimum unaligned end length In the case of targeted data, this is the minimum length of the unaligned end required for a breakpoint to be detected.
- Minimum unaligned end complexity score In the case of targeted data, this is the minimum complexity of the unaligned end required for a breakpoint to be detected. The complexity is based on the Lempel-Ziv algorithm where each unique element in a sequence increases the complexity score by one. For example, if we process the sequence 'ACGGATTC' from left to right, then it has unique elements A, C, G, GA, T, and TC, resulting in a score of 6. Sequences are processed from left to right unless the resulting score is too low, in which case the sequence complexity from right to left is also calculated as this can yield a slightly different score.
- Minimum structural variation score Measure of the overall evidence supporting the structural variant detected. The value is based on the alignment scores of the unaligned ends or, in case of shorter indels, the length of the variation. This value may be increased to reduce the number of structural variants called.
Subsections