Identify Variants (TAS-HD)
You can use the Identify Variants (TAS-HD) ready-to-use workflow to call variants in the mapped and locally realigned reads. The workflow removes false positives and, in case of a targeted experiment, removes variants outside the targeted region. Variant calling is performed with the Fixed Ploidy Variant Detection tool and the Structural Variant Caller.
The Identify Variants (TA-HD) ready-to-use workflow accepts sequencing reads as input.
Run the Identify Variants (TAS-HD) workflow
To run the Identify Variants (TAS-HD) workflow, go to:
Toolbox | Ready-to-Use Workflows | Targeted Amplicon Sequencing (![]() ) | Hereditary Disease (
) | Hereditary Disease (![]() ) | Identify Variants (TAS-HD) (
) | Identify Variants (TAS-HD) (![]() )
)
- Double-click on the Identify Variants (TAS-HD) tool to start the analysis. If you are connected to a server, you will first be asked where you would like to run the analysis.
- Select the sequencing reads you want to analyze (figure 22.74).
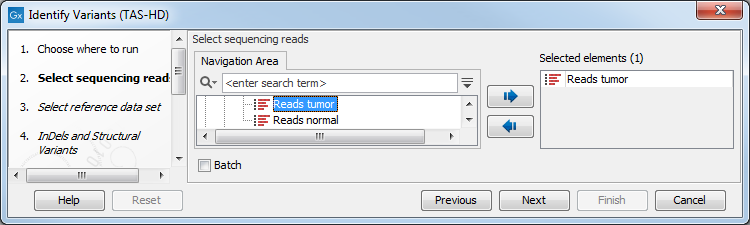
Figure 22.74: Specify the sequencing reads for the sample. - In the next dialog, you have to select which reference data set should be used for the analysis (figure 22.75).
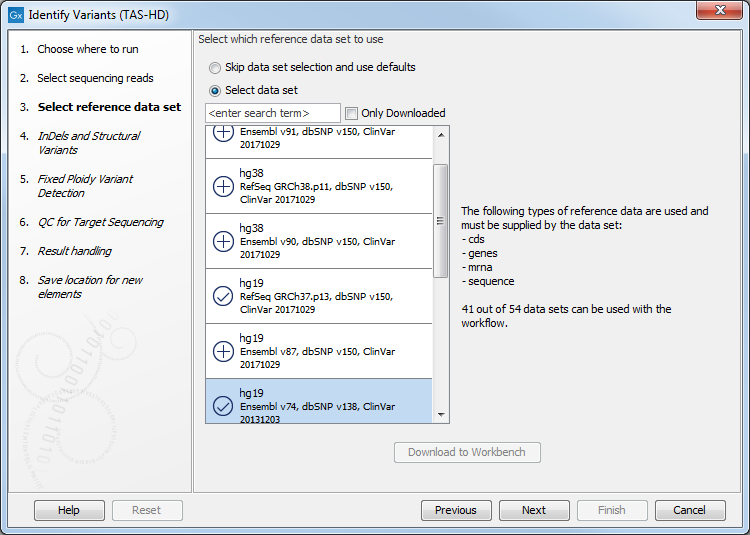
Figure 22.75: Choose the relevant reference Data Set for the analysis. - Specify a target region file for the application (figure 22.76).
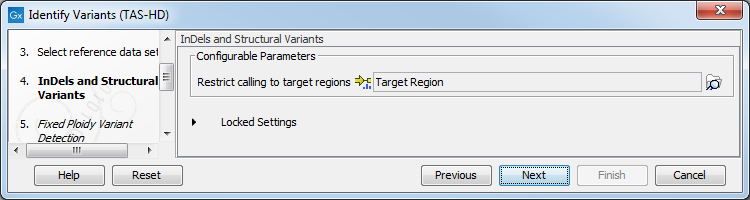
Figure 22.76: Specify the target region file. - Specify the parameters for the Fixed Ploidy Variant Detection tool (figure 22.77).
The parameters used by the Fixed Ploidy Variant Detection tool can be adjusted. We have optimized the parameters to the individual analyses, but you may want to tweak some of the parameters to fit your particular sequencing data. A good starting point could be to run an analysis with the default settings.
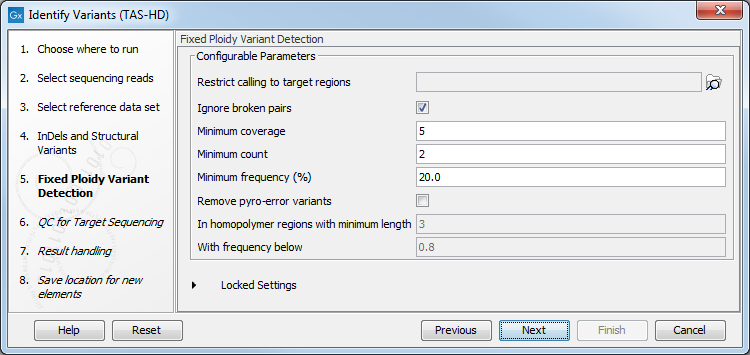
Figure 22.77: Specify the parameters for the Fixed Ploidy Variant Detection tool.The parameters that can be set are:
- Required variant probability is the minimum probability value of the 'variant site' required for the variant to be called. Note that it is not the minimum value of the probability of the individual variant. For the Fixed Ploidy Variant detector, if a variant site - and not the variant itself - passes the variant probability threshold, then the variant with the highest probability at that site will be reported even if the probability of that particular variant might be less than the threshold. For example if the required variant probability is set to 0.9 then the individual probability of the variant called might be less than 0.9 as long as the probability of the entire variant site is greater than 0.9.
- Ignore broken pairs: When ticked, reads from broken pairs are ignored. Broken pairs may arise for a number of reasons, one being erroneous mapping of the reads. In general, variants based on broken pair reads are likely to be less reliable, so ignoring them may reduce the number of spurious variants called. However, broken pairs may also arise for biological reasons (e.g. due to structural variants) and if they are ignored some true variants may go undetected. Please note that ignored broken pair reads will not be considered for any non-specific match filters.
- Minimum coverage: Only variants in regions covered by at least this many reads are called.
- Minimum count: Only variants that are present in at least this many reads are called.
- Minimum frequency: Only variants that are present at least at the specified frequency (calculated as 'count'/'coverage') are called.
- Specify the parameters for the QC for Targeted Sequencing tool (figure 22.78).
When working with targeted data (WES or TAS data), quality checks for the targeted sequencing is included in the workflows. Again, you can choose to use the default settings, or you can choose to adjust the parameters.
The parameters that can be set are:
- Minimum coverage provides the length of each target region that has at least this coverage.
- Ignore non-specific matches: reads that are non-specifically mapped will be ignored.
- Ignore broken pairs: reads that belong to broken pairs will be ignored.
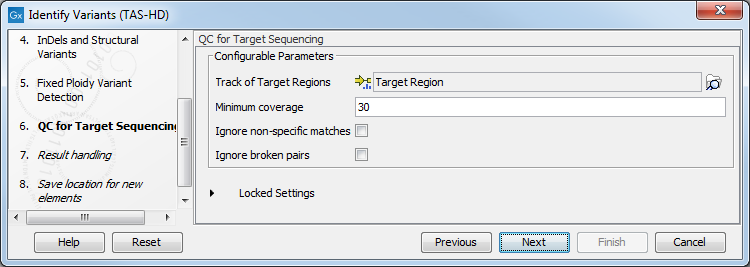
Figure 22.78: Specify the parameters for the QC for Targeted Sequencing tool. - In the last wizard step you can check the selected settings by clicking on the button labeled Preview All Parameters.
In the Preview All Parameters wizard you can only check the settings, and if you wish to make changes you have to use the Previous button from the wizard to edit parameters in the relevant windows.
- Choose to Save your results and click on the button labeled Finish.
Output from the Identify Variants (TAS-HD) workflow
The outputs generated are:
- Read Mapping (
 ) The mapped sequencing reads. The reads are shown in different colors depending on their orientation, whether they are single reads or paired reads, and whether they map unambiguously (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Coloring_mapped_reads.html).
) The mapped sequencing reads. The reads are shown in different colors depending on their orientation, whether they are single reads or paired reads, and whether they map unambiguously (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Coloring_mapped_reads.html).
- Target Regions Coverage (
 ) The target regions coverage track shows the coverage of the targeted regions. Detailed information about coverage and read count can be found in the table format, which can be opened by pressing the table icon found in the lower left corner of the View Area.
) The target regions coverage track shows the coverage of the targeted regions. Detailed information about coverage and read count can be found in the table format, which can be opened by pressing the table icon found in the lower left corner of the View Area.
- Target Regions Coverage Report (
 ) The report consists of a number of tables and graphs that in different ways provide information about the targeted regions.
) The report consists of a number of tables and graphs that in different ways provide information about the targeted regions.
- Identified Variants and Indels Indirect Evidence (
 ) A variant track containing the variants identified by the Fixed Ploidy Variant Detection tool, and a variant track with the indels inferred from indirect evidence by the Structural Variant Caller. The variants can be shown in track format or in table format. When holding the mouse over the detected variants in the Track List, a tooltip appears with information about the individual variants. You will have to zoom in on the variants to be able to see the detailed tooltip.
) A variant track containing the variants identified by the Fixed Ploidy Variant Detection tool, and a variant track with the indels inferred from indirect evidence by the Structural Variant Caller. The variants can be shown in track format or in table format. When holding the mouse over the detected variants in the Track List, a tooltip appears with information about the individual variants. You will have to zoom in on the variants to be able to see the detailed tooltip.
- Genome Browser View Identify Variants (
 ) A collection of tracks presented together. Shows the annotated variant track together with the human reference sequence, genes, transcripts, coding regions, the mapped reads, the identified variants, and the structural variants (see figure 22.33).
) A collection of tracks presented together. Shows the annotated variant track together with the human reference sequence, genes, transcripts, coding regions, the mapped reads, the identified variants, and the structural variants (see figure 22.33).