Identify Ion AmpliSeq SARS-CoV-2 Low Frequency and Shared Variants (Ion Torrent)
The Identify Ion AmpliSeq SARS-CoV-2 Low Frequency and Shared Variants (Ion Torrent) workflow includes all necessary steps for processing the SARS-CoV-2 reads generated with the IonTorrent AmpliSeq pipeline, such as sample QC, filtering of human control reads, variant calling relative to reference MN908947.3 and extraction of a consensus sequence.
The workflow can be found in the Toolbox at:
Ready-to-Use Workflows | SARS-CoV-2 Workflows (![]() ) | Identify Ion AmpliSeq SARS-CoV-2 Low Frequency and Shared Variants (Ion Torrent) (
) | Identify Ion AmpliSeq SARS-CoV-2 Low Frequency and Shared Variants (Ion Torrent) (![]() )
)
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the next step, select the data to be analyzed. These can be sequence lists containing single-end reads selected from the Navigation Area, or by using the "Select files for import" option, where files containing single-end read data can be selected from disk. These will be imported as part of the workflow run. Importing Ion AmpliSeq reads should be done using the Ion Torrent importer.
The workflow contains an Iterate element, allowing each sample to be analyzed individually, before the results are combined for comparison. The "Batch" check box, at the bottom of the dialog, should normally remain unchecked when launching this workflow.
In the next step the relevant Reference Data Set is selected. The workflow uses SARS-CoV-2 reference MN908947.3 by default. Note that alternative reference data sets can be created, as described in http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Custom_Sets.html.
In the next step, you specify how the batch units are defined, that is, which data files come from each individual sample and thus should be analyzed together. Batch units can be defined through the organization of the input data or by using metadata. Further information can be found at http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Running_workflows_in_batch_mode.html. When the metadata option is chosen, selecting an Excel file that describes the data will often be the most convenient method, and it is the only option available when input data will be imported as part of the workflow run. When using already-imported data as input, existing metadata tables, where associations from the input data are already in place, can also be selected.
In the next step, a preview of the batch units is shown. If this looks as expected, you can proceed to configure the analysis settings.
In the next step the Remove False Positives (high frequency) quality filter can be adjusted. High frequency variants (>=50%) with an average base quality lower than the specified value will be discarded (figure 4.4).
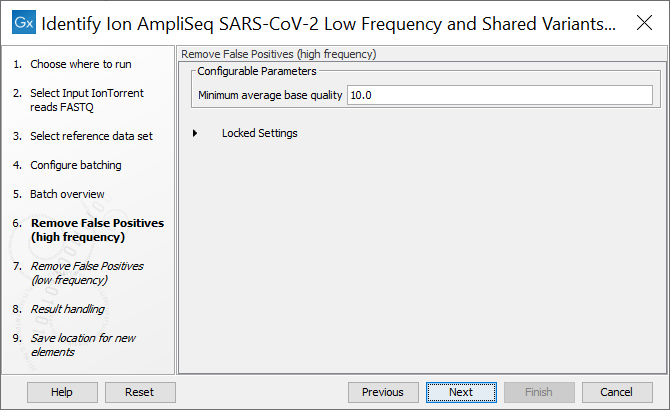
Figure 4.4: The minimum quality for variants to be included in the output track
In the next step, the Remove False Positives (low frequency) quality filter applying filters for both quality and frequency. Lower frequency variants (Default >=20%) with an average base quality lower than the specified value will be discarded by default.
Finally, choose where to save the results.
The outputs generated are described in SARS-CoV-2 workflow output.