Output from the Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina) workflow
The Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina) workflow produces a number of different outputs, some of which are available in a subfolder called Reports and Data. Figure 6.27 shows the structure of the output when looking at the analysis output for one matched tumor-normal pair.
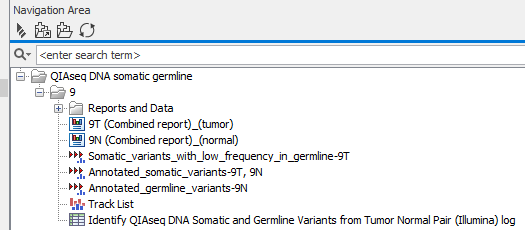
Figure 6.27: The outputs produced by the Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina) workflow. A Track List, three main variant tracks and combined reports for the tumor and the normal sample, respectively are directly accessible, whereas the remaining outputs are placed in a subfolder called Reports and Data.
The outputs generated from the Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina) workflow are:
- Combined_report_tumor (
 ): A combined report for the tumor sample that summarizes information about the most important metrics for the tumor sequencing reads.
): A combined report for the tumor sample that summarizes information about the most important metrics for the tumor sequencing reads.
- Combined_report_normal (): A combined report for the normal sample that summarizes information about the most important metrics for the normal sequencing reads.
- Somatic_variants_with_low_frequency_in_germline (
 ): This variant track holds somatic variants that also are found at a low frequency in the normal sample. More specifically, somatic variants that also are found in more than five normal UMI reads but with a frequency below 20% in the normal sample are reported in this track. For a definition of the variant table content please see
http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=_annotated_variant_table.html.
): This variant track holds somatic variants that also are found at a low frequency in the normal sample. More specifically, somatic variants that also are found in more than five normal UMI reads but with a frequency below 20% in the normal sample are reported in this track. For a definition of the variant table content please see
http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=_annotated_variant_table.html.
- Annotated_somatic_variants (): This variant track holds somatic variants detected in the tumor sample with a variant allele frequency cutoff at 4% and observed in at most five normal reads.
- Annotated_germline_variants (): This variant track holds germline variants detected with a frequency of at least 20% in the normal reads.
- Track List (
 ): A graphic representation that allows for visual inspection of the results. The Track list contains the following collection of tracks:
): A graphic representation that allows for visual inspection of the results. The Track list contains the following collection of tracks:
- Homo sapiens reference sequence
- Reference gene track
- Reference mRNA track
- Per-region_statistics_track_normal
- Per-region_statistics_track_tumor
- Mapped_UMI_reads_normal
- Mapped_UMI_reads_tumor
- Annotated_somatic_variants
- ClinVar variants
- Amino_acid_changes_somatic_variants
- Somatic_variants_with_low_frequency_in_germline
- Annotated_germline_variants
- dbSNP Common
- Amino_acid_changes_germline_variants
- Low_coverage_in_normal
- Low_coverage_in_tumor
- Primer track
- Unfiltered_somatic_variants
- Reports and Data (
 ): A subfolder holding the outputs that are shown in figure 6.28 and described below.
): A subfolder holding the outputs that are shown in figure 6.28 and described below.
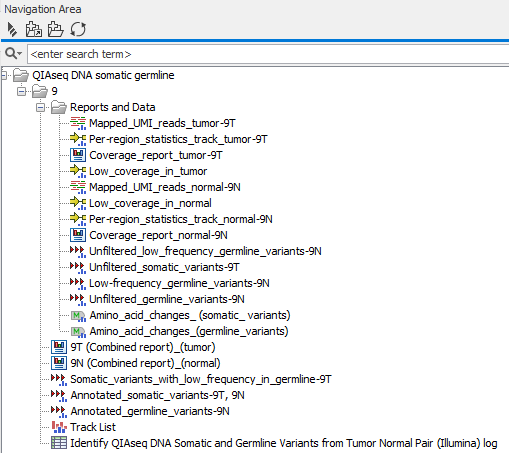
Figure 6.28: All outputs produced by the Identify QIAseq DNA Somatic and Germline Variants from Tumor Normal Pair (Illumina) workflow become visible when unfolding the Reports and Data subfolder.- Mapped_UMI_reads_tumor (
 ): A read mapping of the UMI Reads for the tumor sample.
): A read mapping of the UMI Reads for the tumor sample.
- Per-region_statistics_track_tumor (
 ): A coverage statistics track for the tumor sample with coverage information for each targeted region.
): A coverage statistics track for the tumor sample with coverage information for each targeted region.
- Coverage_report_tumor (): A coverage report for the tumor sample, generated by the QC for Target Sequencing tool (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=QC_Targeted_Sequencing.html).
- Low_coverage_in_tumor (): A coverage track reporting regions with low coverage in tumor. Regions with a coverage below 40x is reported as low-coverage regions.
- Mapped_UMI_reads_normal (): A read mapping of the UMI Reads for the normal sample.
- Low_coverage_in_normal (): A coverage track reporting regions with low coverage in the normal sample. Regions with a coverage below 40x is reported as low-coverage regions.
- Unfiltered_low_frequency_germline_variants (): All detected low-frequency germline variants before applying any filters.
- Unfiltered_somatic_variants (): All detected somatic variants before applying any filters.
- Low_frequency_germline_variants (): Germline variants found with a frequency below 20%.
- Unfiltered_germline_variants (): All detected germline variants before applying any filters.
- Amino_acid_changes_somatic_variants (
 ): A track showing amino acid changes introduced by somatic variants.
): A track showing amino acid changes introduced by somatic variants.
- Amino_acid_changes_germline_variants (): A track showing amino acid changes introduced by germline variants.
- Mapped_UMI_reads_tumor (
The unfiltered variant tracks are provided to allow you to review the raw unfiltered variants. This can be relevant in cases where expected variants are missing from the filtered variants output and potentially have been filtered out due to low quality. The difference between the unfiltered variant track and the variants passing filters track is described in Output from the Identify QIAseq DNA Variants workflows.