Detect Fusion Genes
Detect Fusion Genes is designed to find candidate fusion genes that should typically be investigated further by Refine Fusion Genes, described in Refine Fusion Genes. This tool is generally not run in isolation.
Briefly, the tool works by re-mapping the unaligned ends of reads and determining if these are consistent with a fusion. Fusions are identified from reads that must have an unaligned end close to an exon boundary that can be remapped close to another exon boundary. If the option for Detect fusions with novel exon boundaries has been enabled, the tool also considers reads that are far from an exon boundary and/or whose unaligned ends can be mapped far from an exon boundary in a second pass.
The Detect Fusion Genes tool can be found in the Toolbox at:
Tools | QIAseq Panel Expert Tools (![]() ) | QIAseq RNAscan Panel Expert Tools (
) | QIAseq RNAscan Panel Expert Tools (![]() ) | Detect Fusion Genes (
) | Detect Fusion Genes (![]() )
)
The Detect Fusion Genes tool takes an RNA-seq read mapping as input (figure 7.13).
Figure 7.13: Select an RNA-seq read mapping.
In the next dialog figure 7.14, specify the reference sequence, gene and mRNA track from the CLC_References folder of the Navigation Area. It is possible - but optional - to add a CDS or primer track to run the analysis.
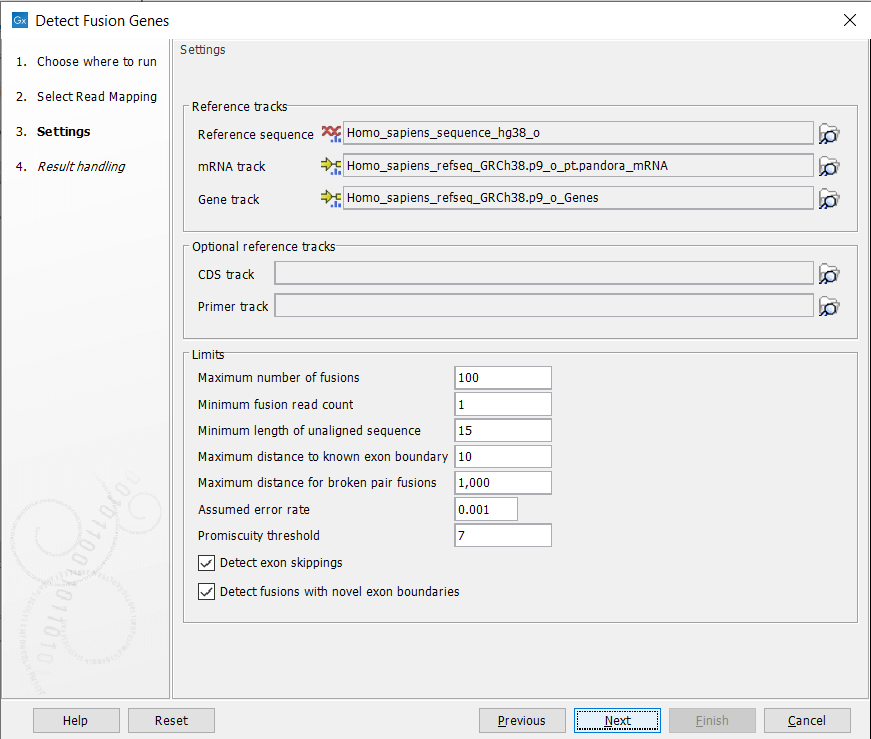
Figure 7.14: Specify references and parameters for the detection.
The additional parameters to set are:
- Maximum number of fusions: The maximum number of putative fusions that will be evaluated. Multiple different possible fusion breakpoints between the same two genes count as 1 fusion.
- Minimum fusion read count: This value is used to calculate Z-score and p-value, by subtracting that number from the total read count before doing the statistics.
- Minimum length of unaligned sequence: Only unaligned ends longer than this will be used for detecting fusions.
- Maximum distance to known exon boundary: Reads with unaligned ends must map within this distance of a known exon boundary, and unaligned ends must map within this distance of another known exon boundary, to be recorded as supporting a fusion event.
Increasing this parameter counts reads that are further from a known exon boundary as if they fused at the boundary, which increases the signal for the fusion. However, increasing the parameter also decreases the resolution at which a fusion can be detected: for example, if "maximum distance to known exon boundary = 10" then two transcripts with exon boundaries 9nt apart will not be distinguished, and the "mRNA (WT + fusions)" output of the tool will only produce one fusion, which can reduce the number of mapping reads that Refine Fusion Genes can use to assess the fusion.
- Maximum distance for broken pairs fusions: The algorithm uses broken pairs to find additional support for fusion events. If a pair of reads originally mapped as a broken pair, but would not be considered broken if mapped across the fusion breakpoints (because the two reads in the pair then get close enough to each other), then that pair of reads supports the fusion event as "fusion spanning reads". The "Maximum distance for broken pairs fusions" parameter specifies how close to each other two broken pairs must map across the fusion breakpoints in order for them to be considered fusion spanning reads. This is usually set to the maximum paired end distance used for the Illumina import of reads.
- Assumed error rate: Value used to calculate Z-score and p-value.
- Promiscuity threshold: Only up to this number of fusion partners will be reported for a given gene.
This parameter does not limit the number of fusion breakpoints that can be reported between two genes, which is capped at 20 pairs of breakpoints:
We limit the number of breakpoint pairs between the same two genes by selecting the highest possible p-value threshold that admits at most 20 breakpoint pairs.
- Detect exon skippings: Check this option to consider exon skippings.
- Detect fusions with novel exon boundaries: When enabled, fusions beyond the distance set for "Maximum distance to known exon boundary" are additionally reported where breakpoints are not at canonical exon boundaries.
In the Result handling dialog, it is possible to choose to output a report with unaligned ends information (figure 7.15):
- Unaligned ends: number of found unaligned ends.
- Mapped unaligned ends: number of unaligned ends which could be mapped
- Unmapped unaligned ends: number of unaligned which could not be mapped.
- Discarded base breakpoints: when two transcripts of the same gene overlap so that two breakpoints are found next to each other, one of them will be discarded.
This report can be used together with the Combine Reports tool (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Combine_Reports.html)

Figure 7.15: Unaligned ends report.
Known limitations
- The tool is not suitable for detection of circRNAs. Evidence of back-splicing is filtered out.
- Fusions that involve a mix of sense and antisense exons are filtered out.
- Fusions that involve more than two genes in the fusion product are not explicitly detected.
- Fusions will not be reported for a gene if they involve fusing into a region before the first annotated exon or after the last annotated exon of that gene.
Detect Fusion Genes otherwise generates a read mapping, an unaligned ends track, a fusion track (see the details of the fusion track in Fusion tracks, and several tracks for use in Refine Fusion Genes. The unaligned ends track is useful when choosing how to set the parameters "Minimum fusion read count", "Minimum length of unaligned sequence", and "Maximum distance to exon boundary" for a particular panel and sequencing protocol in order to find known fusions, as it shows which unaligned ends of reads were considered and where they were mapped.