Calculate Unique Molecular Index Groups
The tool Calculate Unique Molecular Index Groups annotates the mapped reads with a "Unique Molecular Index group ID", that is identical for reads that are determined to belong to the same UMI. The tool can be found in the Toolbox at:
Tools | QIAseq Panel Expert Tools (![]() ) | QIAseq DNA Panel Expert Tools (
) | QIAseq DNA Panel Expert Tools (![]() ) | Calculate Unique Molecular Index Groups (
) | Calculate Unique Molecular Index Groups (![]() )
)
In the first dialog (figure 6.59), select a read mapping of reads that were previously annotated with UMI annotations.
Figure 6.59: Select a read mapping made from reads whose UMI was removed and annotated on the sequences.
The grouping of reads into UMI groups works as follows:
- The tool groups reads that
- start at the same position based on the end of the read to which the UMI is ligated (i.e., Read2 for paired data),
- are from the same strand, and
- have identical UMIs.
- The tool then fuzzy merges singleton groups into non-singleton groups, if the UMI of the singleton group can be made into UMI of non-singleton group by introducing a SNP, and if the non-singleton group is the biggest of such group (i.e., if two different introduced SNPs yields two different non-singleton groups, the biggest one is chosen).
- Additional merging of singletons and small groups into bigger ones can happen depending on the parameters set for the tool.
It is possible to change the following parameters (figure 6.60):
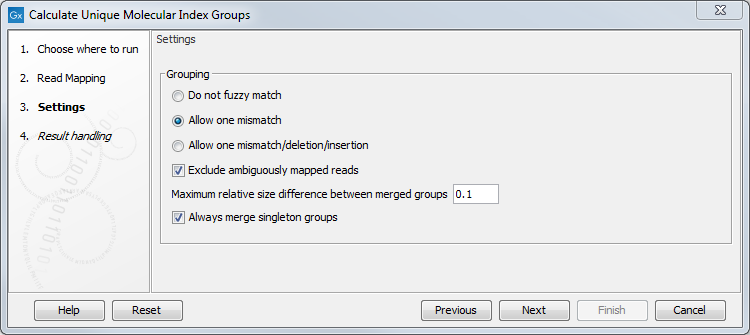
Figure 6.60: Select a read mapping made from reads whose UMI was removed and annotated on the sequences.
- Do not fuzzy match Groups will be merged only if they match exactly.
- Allow one mismatch Groups will be merged if they are at most one mismatch apart.
- Allow one mismatch/deletion/insertion Groups will be merged if they are at most one mismatch, deletion or insertion apart.
- Exclude ambiguously mapped reads is checked by default.
- Maximum relative size difference between merged groups will merge small groups into bigger ones if the size difference between the two groups is smaller than a certain value (set at 0.1 per default).
- Always merge singleton groups When this option is checked, a singleton UMI group is merged with a non-singleton group even if the "Maximum relative size difference between merged groups" threshold is not met.
Click Next to choose whether to Open or Save the resulting read mapping of reads which now have had a "UMI group ID" annotation.
A report can also be generated. It contains:
- A table with the following information:
- Reads in input: reads that were aligned to the reference
- Reads mapped multiple places (discarded): reads that aligned to the reference in multiple places, and thus discarded
- Groups merged
- Groups not merged due to >1 candidate of equals size
- Groups not merged due to parameter thresholds
- Number of groups that were too small (discarded)
- Number of reads in groups that were too small (discarded)
- Output groups
- Singleton groups
- Reads in largest group
- Number of Unique Molecular Indices in most divergent group
- Average, Median and Standard deviation of reads per group
- Reads by group size of 25th, 50th and 75th percentile. Size is the number of reads in a group.
- Groups with sizes <=x (% of groups) (% of reads): A series of values reporting the number of groups containing up to a particular number of reads, followed by the percentage of UMI groups this represents and the percentage of all reads included in these groups.
- The following plots are also available:
- Reads by group size. Showing the number of reads in group by group sizes. The second plot includes only groups with fewer than 50 reads
- Group Sizes graphs. The first plot shows the sizes of all UMI groups. The second includes the sizes of only groups with fewer than 50 reads.
Note: When the group sizes (the number of reads in UMI groups) are very large (in most cases more than 10 reads in a UMI group is not desirable), this can indicate problems, such as quality issues with the sample. It can also indicate that the sequencing depth could be reduced.