Identify DNA Germline Variants workflow
The Identify DNA Germline Variants workflow includes the minimum analysis steps necessary for variant detection when starting with a set of sequencing reads. This workflow also includes steps to generate reports and visualizations.
We recommend that samples with known variants are used to test and to optimize the workflow settings to fit your specific application. Suggestions for customizations of this workflow are provided at the end of this section.
The workflow requires reference data. Any available, relevant reference sequence can be chosen. Reference data for some organisms can be downloaded using the Reference Data Manager (References management).
Launching the workflow
The Identify DNA Germline Variants workflow is at:
Toolbox | Template Workflows | Basic Workflow Designs (![]() )
| Identify DNA Germline Variants (
)
| Identify DNA Germline Variants (![]() )
)
Launch the workflow and step through the wizard.
- Select the sequence lists containing the reads to analyze and click on Next.
- Select a reference data set or select "Use the default reference data" if you want to specify individual reference elements in the next wizard step (figure 12.67).
- Specify the trimming settings.
- If the data is from a targeted sequencing experiment, you can restrict the InDels and Structural Variants tool to call variants only in target regions by providing a target regions track (figure 12.68).
- If the data is from a targeted sequencing experiment, you can restrict Fixed Ploidy Variant Detection to call variants only in target regions by providing a target regions track.
- Specify thresholds for the variants to be reported (figure 12.69).
- Finally select a location to save outputs to and click on Finish.
Tools in the workflow and outputs generated
The tools and outputs provided by this workflow are:
- QC for Sequencing Reads performs basic QC on the sequencing reads and outputs a report that can be used to evaluate the quality of the sequencing reads. See QC for Sequencing Reads. Here the report is included in a combined report, together with other reports, rather than being output as a separate report.
- Trim Reads trims reads for adapter sequences and low quality nucleotides. The appropriate settings for Trim Reads depend on the protocol used to generate the reads.
See NGS Trim Reads tool for more details about the Trim Reads tool. One of the outputs of this tool is a report, which here is included in a combined report, together with other reports, rather than being output as a separate report.
- Map Reads to Reference maps reads to a reference sequence. See Map Reads to Reference.
- Indels and Structural Variants is used to predict InDels. Identified InDels are used to improve the read mapping during a realignment step. The identified InDels are also output to a track called Indels-indirect_evidence. With the default settings, only reads with 3 or fewer mismatches to the reference are considered when identifying potential breakpoints from unaligned ends. This may need to be adjusted if long and/or low quality reads are used as input. See InDels and Structural Variants.
- Local Realignment uses the predicted InDels from Indels and Structural Variants to realign the reads and hence improve the read mapping Local Realignment. The resulting Reads Track is output with the name Mapped_reads.
- Fixed Ploidy Variant Detection calls variants in the read mapping that are present at germline frequencies. In this workflow, the coverage threshold for variant detection is set to 10, meaning that no variants will be called in regions where the coverage is below 10. Similarly, a frequency threshold of 20 percent has been defined. See section Fixed Ploidy Variant Detection.
- Remove Marginal Variants allows post-filtering of detected variants based on frequency, forward/reverse balance and minimum average base quality. See Remove Marginal Variants. The tool outputs the final variant list, called Variants_passing_filters. Note that decreasing the thresholds in this tool below the thresholds set in Fixed Ploidy Variant Detection will not result in detection of additional variants.
- Create Track List outputs a track list Genome_browser_view containing the reference sequence, read mapping and identified variants. See Track lists as workflow outputs.
- Create Sample Report compiles the reports from other tools and outputs a Combined_report. It is possible to set QC thresholds in this tool, which results in an additional section in the report showing whether QC thresholds were met (see Report types supported).
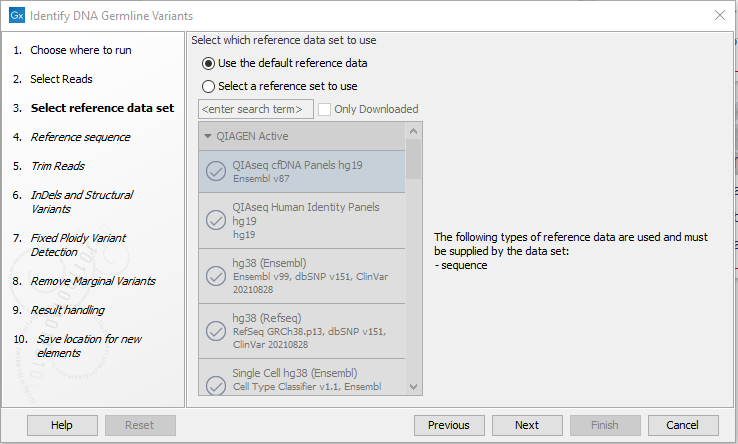
Figure 12.67: Wizard step for specifying reference data.
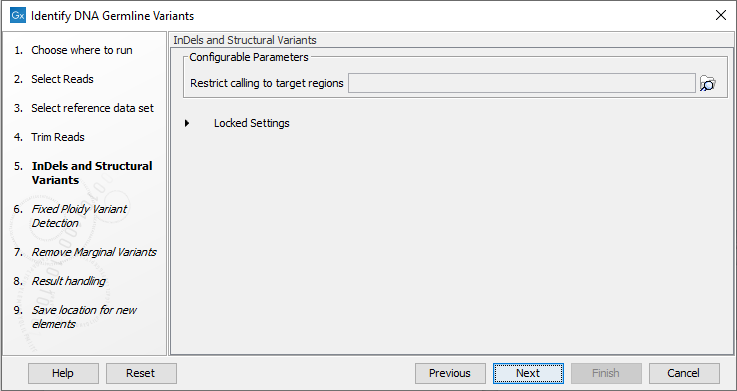
Figure 12.68: Optional wizard step for selecting a target region.
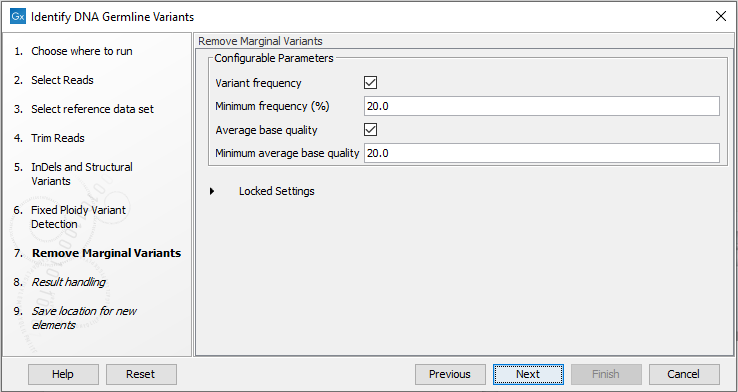
Figure 12.69: Wizard step for specifying variant filtering criteria.
Customizing the Identify DNA Germline Variants workflow
Template workflows can be easily edited to add or remove analysis steps, change parameter settings, and so on. See Template workflows for information about how to open a template workflow for editing.
Some changes that may be of particular interest when working with the Identify DNA Germline Variants workflow are:
- Low frequency variant detection To detect low frequency variants, the Fixed Ploidy Variant Detection workflow element should be replaced with Low Frequency Variant Detection (section Low Frequency Variant Detection).
- Targeted sequencing
- Several tools in this workflow can be configured to consider only defined target regions, which is useful if a targeted protocol was used to generate the sequencing data. Making this adjustment typically reduces the runtime of the analysis substantially. This change can be made by editing the workflow, but you can also select target regions in the wizard when launching the template workflow.
- Adding the tool QC for Targeted Sequencing to the workflow can also be useful. This generates a report with useful metrics, such as the percentage of reads mapped in the target regions QC for Targeted Sequencing.
- PCR duplicates For protocols where PCR bias is expected, it can be useful to remove PCR duplicates from the read mapping. This can be achieved with the tool Remove Duplicate Mapped Reads (Algorithm details and parameters). For inspiration, take a look at the workflow Identify Variants (WES-HD) distributed with the Biomedical Genomics Analysis plugin https://resources.qiagenbioinformatics.com/manuals/biomedicalgenomicsanalysis/current/index.php?manual=Identify_Variants_WES_HD.html.
- Annotation of variants Variants can be annotated with different types of information, such as the gene they occur in and whether the variant changes the coding sequence of a protein. For inspiration, see the workflow Annotate Variants (WGS) distributed with the Biomedical Genomics Analysis plugin https://resources.qiagenbioinformatics.com/manuals/biomedicalgenomicsanalysis/current/index.php?manual=Annotate_Variants_WES.html.
- Filtering of variants
Variants can be filtered according to information in their annotations, such as their quality and their position relative to a gene (see Variant filtering).
For inspiration, you can refer to template workflows distributed with the Biomedical Genomics Analysis plugin, where many include filtering cascades aimed at removing false positive variants from the final variant list produced. Examples include the Identify QIAseq DNA Variants workflows (see https://resources.qiagenbioinformatics.com/manuals/biomedicalgenomicsanalysis/current/index.php?manual=_Identify_QIAseq_DNA_Variants_ready_to_use_workflows.html).