The clc_remove_duplicates Program
clc_remove_duplicates is designed to filter out duplicate reads, retaining just a single representative of any set of identical reads in the output. It is designed for use in situations where certain regions are represented by a higher numbers of reads than reflects the relative underlying biological abundance, such as can occur using PCR amplification. The challenge is to achieve this without removing reads that represent the true biological situation, e.g. repeat regions.
When to use and when not to use this tool
We believe this tool should be rarely needed in practice and we recommend it is only used in situations where there is a strong suspicion that duplicate reads are present and that they would affect the quality of a de novo assembly. We note that NGS reads often contain leftover adapters and sequencing artifacts, and that these can cause a massive increase in both memory and time consumption of this tool. We thus do not recommend that this tool in included as a fixed step in assembly pipelines.
This tool is also not recommended for use with for any data where the start of a large number of reads is expected to be the same location on a genome, for example exome data, amplicon data, RNA-Seq data, or Chip-Seq data. It should also not be used with data that will be used for variant detection downstream.
Recommendations when running this tool
- clc_remove_duplicates should be run after adapters have been trimmed from the reads.
- Data should not be trimmed based on quality before running this tool.
clc_remove_duplicates initially looks for identical words at the beginning of reads. Thus, if adapters are present, many reads will be falsely identified as duplicates. Conversely, if reads have been trimmed based on quality, true duplicates may not be identified as they may have different start points after trimming.
What is a duplicate read?
An example of true duplicate reads can be seen in figure 8.1, where the reads have been mapped to a reference sequence. The duplicates can be easily seen: they map to the same position on the reference with the same mapping orientation, and there is a sudden rise in coverage at that point.
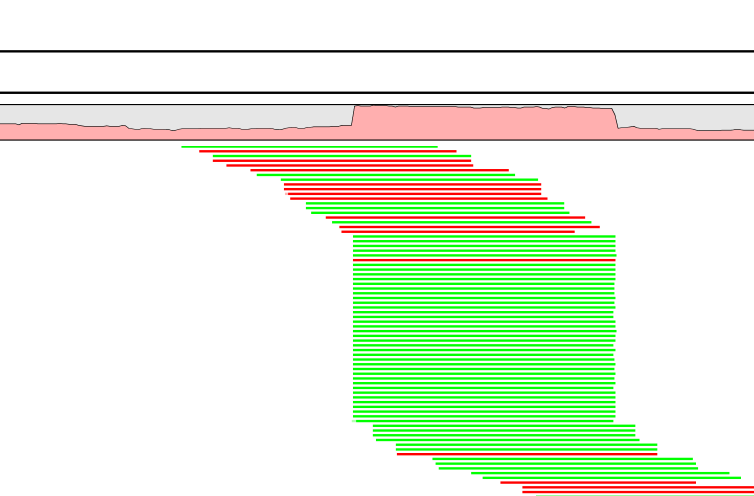
Figure 8.19: Duplicate reads in this mapping are easily identified: they map with the same start position and in the same orientation (all colored green, showing they map to the forward strand of the reference). The region they map to has an unpectedly high coverage.
In a data set without duplicate reads, there are still fluctuations in coverage, but a large number of reads do not start at exactly the same position with identical orientation. An example is shown in figure 8.2.
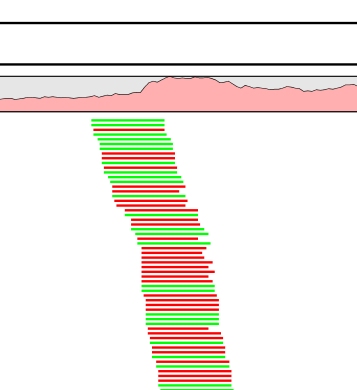
Figure 8.20: A rise in coverage is seen in this region, but this does not appear to be due to duplicate reads.
Looking for neighbors
clc_remove_duplicates works directly on the sequencing reads to identify duplicates by looking for "neighboring" reads, that is, reads that share most of the sequence but with a small offset. These are used to determine whether there is generally high coverage for this sequence. If there is not, the read in question will be marked as a duplicate.
For certain sequencing platforms such as 454, reads have varying lengths. This is taken into account by the algorithm.
Sequencing errors in duplicates
clc_remove_duplicates accounts for sequencing errors when it identifies duplicate reads. This is done by defining the limits of how different reads can be and still be considered duplicates.
Reads can be considered duplicates if:
- They share enough common sequence: single reads share a common sequence of at least 20 bases at the start, or at any of four other regions distributed evenly across the read. For paired reads, the length of common sequence required is 10 bases.
- The remainder of the read has an alignment score above 80% of the optimal score, where the optimal score is what a read would score if it aligned perfectly to the consensus for that group of duplicates.
An illustration of the problem these checks are addressing: If a set of reads contained 100 duplicates of a particular 100bp read, and ther was a random 0.1 % probability of a sequencing error, 10 of those duplicates would, on average, contain an error. Without accounting for this, only the 90 identical reads would be removed, leaving the 10 duplicate reads with errors in the output.
Paired data
For paired reads, we check whether the sequences in different pairs mapping in a given region have their first 10 base pairs identical. If they do, they are marked as potential duplicate reads. (We have found this route works better than a statistical route in the case of paired data.) We then check our proposed list of duplicates to look for false positives. We also do a check for closely related variants of those sequences we now believe to represent duplicate reads.
The algorithm also takes sequencing errors into account when filtering out paired data.
Known limitations
clc_remove_duplicates has a limitation when there are duplicate reads representing several alleles. The algorithm will identify if there are duplicate reads to be removed, but it is not able to distinguish between sequencing errors and true variation in the reads. So if you have a heterozygous SNP in such an area, you risk that only one of the alleles will be represented in the data after running this tool.
Subsections