Method
Like our old assembler, the long read assembler builds a kind of de Bruijn graph to do the assembly.
Since the method is ![]() -mer based, a (fixed) word size
-mer based, a (fixed) word size ![]() , should be chosen and provided to the assembler using the
, should be chosen and provided to the assembler using the -w/--wordsize parameter. If left out, the assembler will do its best to decide on a word size based on the size of the input data.
When the assembler is run, we first count all the ![]() -mers occurring in the reads, provided as one or more FASTA or FASTQ files containing the reads. The
-mers occurring in the reads, provided as one or more FASTA or FASTQ files containing the reads. The ![]() -mers are identified with their reverse-complements as we have no way of knowing which strand gave rise to a particular read.
-mers are identified with their reverse-complements as we have no way of knowing which strand gave rise to a particular read.
Once all the ![]() -mers have been counted, we throw away all
-mers have been counted, we throw away all ![]() -mers that occur fewer than a user-specified number of times, since unique, or rarely occurring,
-mers that occur fewer than a user-specified number of times, since unique, or rarely occurring, ![]() -mers can often be attributed to sequencing errors.
-mers can often be attributed to sequencing errors.
In the next step, any ![]() -mers that always occur next to each other, i.e. only have
-mers that always occur next to each other, i.e. only have ![]() overlap with one another, are joined together, iteratively, into longer fragments5.4.
overlap with one another, are joined together, iteratively, into longer fragments5.4.
Next, we do a second pass through the read data; mapping all the reads back to the fragments. Reads that span/link multiple fragments give rise to connections between the fragments, and together, the fragments (nodes) and those connections (edges) form a graph structure. If there is a gap between two fragments, that gap is replaced by a majority consensus sequence of the reads spanning it.
From this point, the assembly graph represents our knowledge of the genome that we aim to assemble. However, it is often a very messy graph, and it is not immediately possible to read off the chromosome, or chromosomes, that we are trying to assemble. Luckily, we have a lot of information available, such as information about how well the individual edges are supported. We use this information along with the graph topology itself to reduce noise and tease out the actual information content of the graph. This simplification process consists of many, quite technical, steps and will not be covered in any detail here.
Once the graph has been simplified into a number of longer contigs, as an optional step, it is possible to polish the contigs by mapping the reads back onto the contigs, replacing them with the respective consensus sequences. In the case of error-corrected PacBio reads, you will get the best result by using the original raw reads, instead of the error-corrected ones, to do the polishing.
Finally, the resulting assembly/contigs are output to a FASTA file.
Footnotes
- ...fragments5.4
- Given a set of reads, word size
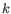 , and minimum coverage
, and minimum coverage 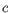 ; the resulting fragments are unambiguously defined.
; the resulting fragments are unambiguously defined.