OTU clustering parameters
After having selected the sequences you would like to cluster, the wizard offers to set some general parameters (see figure 5.3).
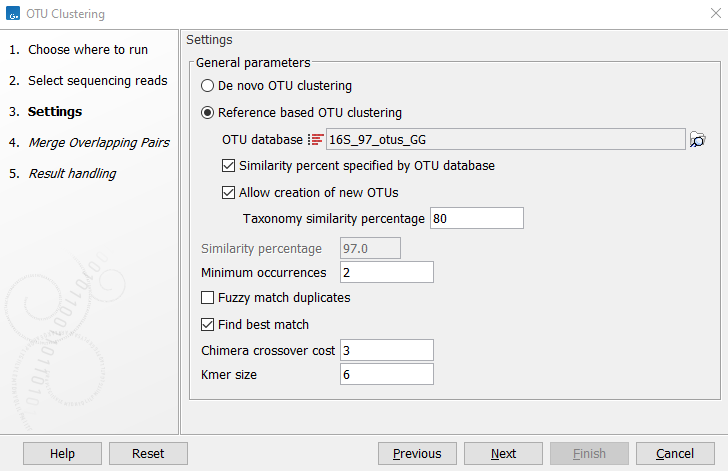
Figure 5.3: Settings for the OTU clustering tool.
You can choose to perform a De novo OTU clustering, or you can perform a Reference based OTU clustering.
The following parameters can be set:
- OTU database Specify the reference database to be used for Reference based OTU clustering. Reference databases can be created by the Download Amplicon-Based Reference Database tool or the Update Sequence Attributes tool.
- Similarity percent specified by OTU database Will apply the same similarity percentage (see below) as what was used when creating the reference database. This parameter is available only when performing a reference based OTU clustering. Selecting this parameter will disable the similarity percent parameter.
- Allow creation of new OTUs Allows sequences which are not already represented at the given similarity distance in the database to form a new cluster, and a new centroid is chosen. This parameter can be set only when performing a "Reference-based OTU clustering". Disallowing the creation of new OTUs is also known as closed reference OTU picking. Note that for input data where reads do not have the same orientation, the direction of the new OTUs cannot be inferred consistently. This may cause problems in downstream analyses (e.g. for estimating phylogenetic diversity).
- Taxonomy similarity percentage Specifies the similarity percentage to be used when annotating new OTUs. This parameter is available only when Allow creation of new OTUs is selected.
- Similarity percentage Specifies the required percentage of identity between a read and the centroid of an OTU for the read to join the OTU cluster.
- Minimum occurrences Specifies the minimum number of times a sequence must be represented in the read set for it to be included in the analysis. A value of 2 means that at least two reads representing a given sequence (i.e. duplicates) must be present for that sequence to be represented in further analysis. This option can be useful for filtering out singletons.
- Fuzzy match duplicates Specifies how to define duplicate reads. When not selected, reads that are 100% identical are considered duplicates. When selected, reads with 2% or fewer single nucleotide differences between them, and no other differences, are considered duplicates. The reads are sorted lexicographically (dictionary order) and then processed from most abundant to the least. Using this option, two or more singleton reads that are very similar may be marked as duplicates, allowing them to be included in further processing if together, their number exceeds the "Minimum occurrences" value.
- Find best match If not selected, a read becomes a member of the first OTU-database entry found within the specified threshold. If the option is selected all database entries are tested and the read becomes a member of the best matching result. Note that "first" and "all" are relative terms in this case as kmer-searches are used to speed up the process. "All" only includes the database entries that the kmer search deems close enough, i.e., database entries that cannot be within the specified threshold will be filtered out at this step. "First" is the first matching entry as returned by the kmer-search which will sort by the number of kmer-matches.
- Chimera crossover cost The cost of doing a chimeric crossover, i.e. the higher the cost the less likely it is that a read is marked as chimeric.
- Kmer size: The size of the kmer to use in regards to the kmer usage in finding the best match.
Chimera detection is performed as follows: The read being processed is split into fragments. Each fragment is then queried for matches against the database with a k-mer search. Database references that match at least one query fragment are then selected and the read is then aligned to each selected reference while allowing "crossovers". Chimera detection is performed in order to identify any chimeric sequences, i.e., amplicons formed by joining two sequences during PCR. These are artifacts that will be excluded from the regular OTU clustering, and presented in a different abundance table labeled as being chimera-specific.
In order to use the highest quality sequences for clustering, it is recommended to merge paired read data. If the read length is smaller than the amplicon size, forward and reverse reads are expected to overlap in most of their 3' regions. Therefore, one can merge the forward and reverse reads to yield one high quality representative according to some pre-selected merge parameters: the overlap region and the quality of the sequences. For example, for a designed 150 bp overlap, a maximum score of 150 is achievable, but as the real length of the overlap is unknown, a lower minimum score should be chosen. Also, some mismatches and indels should be allowed, especially if the sequence quality is not perfect. You can also set penalties for mismatch, gap and unaligned ends.
In the Merge Overlapping Pairs dialog, you can set the parameters as seen in figure 5.4.
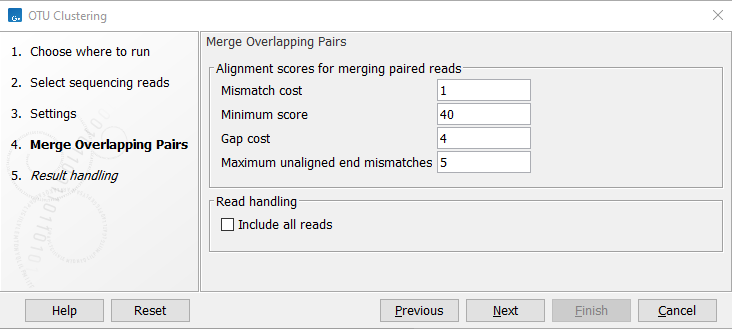
Figure 5.4: OTU Clustering parameters for merging of overlapping pairs.
In order to understand how these parameters should be set, an explanation of the merging algorithm is needed: Because the fragment size is not an exact number of base pairs and is different from fragment to fragment, an alignment of the two reads has to be performed. If the alignment is good and long enough, the reads will be merged. Good enough in this context means that the alignment has to satisfy some user-specified score criteria (details below). Because of sequencing errors that typically are more abundant towards the end of the read, the alignment is not expected always to be perfect, and the user can decide how many errors are acceptable. Long enough in this context means that the overlap between the reads has to be non-coincidental. Merging two reads that do not really overlap leads to errors in the downstream analysis, thus it is very important to make sure that the overlap is big enough. If only a few bases overlap was required, some read pairs will match by chance, so this has to be avoided.
The following parameters are used to define what is good enough and long enough.
- Mismatch cost The alignment awards one point for a match, and the mismatch cost is set by this parameter. The default value is 1.
- Minimum score This is the minimum score of an alignment to be accepted for merging. The default value is 40. As an example: with default settings, this means that an overlap of 43 bases with one mismatch will be accepted (42 matches minus 1 for a mismatch).
- Gap cost This is the cost for introducing an insertion or deletion in the alignment. The default value is 4.
- Maximum unaligned end mismatches: The alignment is local, which means that a number of bases can be left unaligned. If the quality of the reads is dropping to be very poor towards the end of the read, and the expected overlap is long enough, it makes sense to allow some unaligned bases at the end (the default value is 5). However, this should be used with great care: a wrong decision to merge the reads leads to errors in the downstream analysis, so it is better to be conservative and accept fewer merged reads in the result. Please note that even with the alignment scores above the minimum score specified in the tool setup, the paired reads also need to have the number of end mismatches below the "Maximum unaligned end mismatches" value specified in the tool setup to be qualified for merging.
The tool accepts both paired and unpaired reads but will only merge paired reads in forward-reverse orientation. After merging, the merged reads will always be in the forward orientation.
- Include all reads Select this option to include the non-merged reads in the OTU clustering analysis. If some or most of your paired reads are not expected to overlap, you should check this option to include all reads in the analysis. An example of an application resulting in non-overlapping paired reads would be fungal ITS sequencing, where you often sequence a larger amplicon than what can be covered by your read pairs.