Find Prokaryotic Genes
The Find Prokaryotic Genes tool allows you to annotate a DNA sequence with CDS information. The tool is currently for use with near-complete single prokaryotic genomic and metagenomic data.
The tool creates a gene prediction model from the input sequence, which estimates GC content, conserved sequences corresponding to ribosomal binding sites, start and stop codon usages, and a statistical model (namely, an Interpolated Markov Model) for estimating the probability of a sequence to be part of a gene compared to the background. The model is then used to predict coding sequences from the input sequence. Note that this tool is inspired by Glimmer 3 (see https://ccb.jhu.edu/papers/glimmer3.pdf).
To maximize the gene prediction accuracy, the gene models should be trained on sequences that belong to the same species or to similar ones. When the input consists of sequences originating from multiple organisms, it is recommended to build a gene model for each organism by choosing the "Learn one gene model for each assembly" option. In the Assembly grouping parameters, there are multiple options for specifying what should be considered an assembly.
To start the analysis, go to:
Tools | Microbial Genomics Module (![]() ) | Functional Analysis (
) | Functional Analysis (![]() ) | Find Prokaryotic Genes (
) | Find Prokaryotic Genes (![]() )
)
In the first dialog, select input sequences. The input should consist of one or few contigs from the same species. If several sequences are provided as input, the model training can be used to specify if the tool should build a separate model for each assembly. The tool can also be run in batch mode.
In the second dialog (figure 12.1), it is possible to configure the tool.
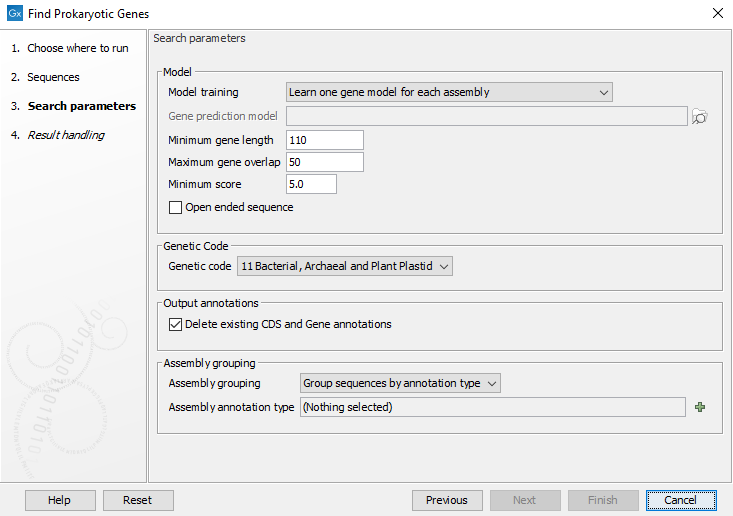
Figure 12.1: Configuring the Find Prokaryotic Genes tool
- Model
- Learn one gene model: Learns a single model from the data. Assumes that the sequences come from one organism or a group of closely related organisms.
- Learn one gene model for each assembly: Learns a model for each assembly or bin. This option should be used when assemblies can be clearly distinguished, for example when they are separated with one assembly per sequence list or contain the "Assembly ID" attribute as is the case with output from Bin Pangenomes by Taxonomy, Download Custom Microbial Reference Database and the prokaryotic databases from Download Curated Microbial Reference Database.
- Use a previously trained model and use its default parameters: This option allows to choose a model that has been previously trained and run the analysis with the same parameters used when training the model.
- Use a previously trained model: This option allows to choose a model that has been previously trained. It also allows to modify some parameters.
In all but one of this option, the following parameters can be modified:
- Minimum gene length: in bp, excluding start and stop codons.
- Maximum gene overlap: in bp
- Minimum score: Putative genes with a score below this value will be ignored. The value of a gene score depends on how well the sequence of the gene matches the model. It is computed by taking into account how much the sequence is typical of a coding region (as opposed to background noise or the same coding region read in a different frame), of the prevalence of the start codon, and of the presence of a putative ribosomal binding site near the start codon.
- Open ended sequence: check this option to annotate open-ended sequences, which is particularly useful for annotating small contigs.
- Genetic Code
The genetic code to use (default to bacterial). This genetic code is used to determine which stop codons should be used and to compute a background distribution for amino-acid usage.
- Output annotations
Delete Existing CDS and Gene Annotations. This is selected by default in order to avoid having many duplicate annotations. Unchecking is useful if one wants to compare the results with other annotations.
- Assembly Grouping
- Each sequence is one assembly: Each individual sequence is considered a complete assembly of a genome.
- Each input element is one assembly: Each input element, i.e. input sequence or input sequence list, is considered a complete assembly of a genome.
- Group sequences by annotation type: Use annotations to group the assemblies and specify the annotation field with Assembly annotation type. Some tools, such as the Download Custom Microbial Reference Database, will automatically assign an "Assembly ID" that can be used for grouping. For manual assignment, please see Using the Assembly ID attribute.
The tool will output a copy of the input sequence with CDS and Gene annotations. It is possible to save the gene model(s) used for the analysis when the option "Learn one gene model" was selected earlier. This model can then be reused to annotate other input sequences by setting the "Model Training" option to "Use a previously trained model" or "Use a previously trained model and use its default parameters".