Create SNP Tree
The Create SNP Tree tool is inspired by [Kaas et al., 2014].
To generate a SNP tree, first map reads from the individual samples to a common reference and call variants. The corresponding tools are described at:
- Map Reads To Reference: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Mapping_parameters.html
- Variant detection: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Variant_detection.html
To create a SNP tree, go to:
Toolbox | Microbial Genomics Module (![]() ) | Typing and Epidemiology (
) | Typing and Epidemiology (![]() ) | Create SNP Tree (
) | Create SNP Tree (![]() )
)
In the first dialog, select reads tracks or read mappings (figure 9.1).
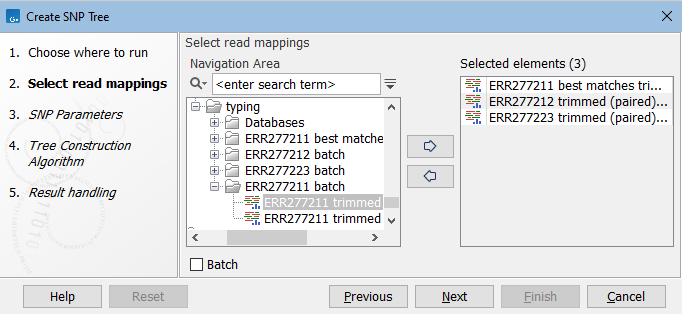
Figure 9.1: Select read mappings to be included in the SNP tree analysis.
Next, select Variant parameters. These determine which SNPs (single-nucleotide polymorphisms) and MNVs (multi-nucleotide variants) to consider for building the SNP tree:
- Variant track. Select variant tracks that correspond to the previously selected reads tracks or read mappings (figure 9.2). The variant tracks determine which positions to potentially include in the SNP tree.
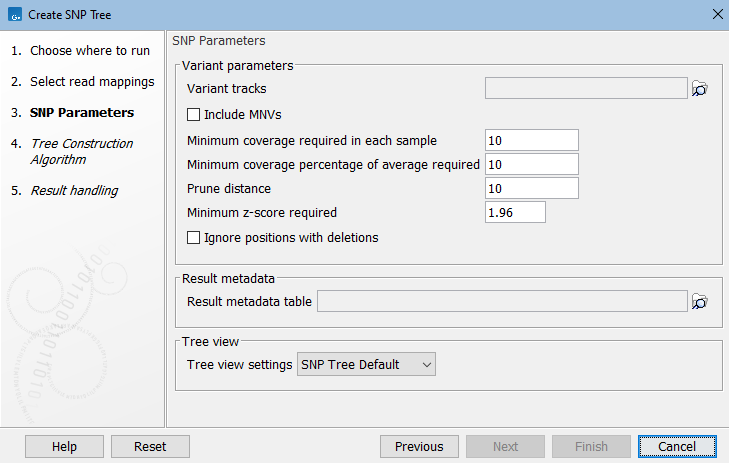
Figure 9.2: Select variant tracks and specify relevant parameters before generation of a SNP tree. - Include MNVs. Check this option to include MNVs along with SNPs when building the SNP tree.
- Minimum coverage required in each sample. Positions are filtered if at least one sample has coverage below the specified threshold.
- Minimum coverage percentage of average required. Positions are filtered if the coverage of at least one sample falls below the specified percentage of the average coverage of that sample.
- Prune distance. Minimum number of nucleotides between unfiltered positions. If a position is within this distance of a previously used position it will be filtered.
- Minimum z-score required. Defining
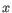 as the number of the most prevalent nucleotide at a position and
as the number of the most prevalent nucleotide at a position and 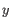 as the coverage subtracting , the z-score is calculated as
as the coverage subtracting , the z-score is calculated as
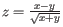 . If the calculated z-score for a given position is less than the specified minimum value the position is filtered.
. If the calculated z-score for a given position is less than the specified minimum value the position is filtered.
- Ignore positions with deletions. Check this option to ignore SNP positions where at least one sample has a deletion at the given position.
The initial list of SNP positions is reduced based on the above filters. Of the remaining, only variants with relative frequency above 50% (haploid organisms) will be considered. Information about reference and alleles is deduced from the read mappings.
Select the Result metadata table with metadata relevant for your samples. This will allow you to decorate the resulting SNP tree with metadata information, see SNP tree.
Select Tree view settings. (None, K-mer Tree Default, or SNP Tree Default) or your own custom tree setting. Read more on tree settings in general at https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Tree_Settings.html.
In the next dialog, select the tree construction algorithm (figure 9.3).
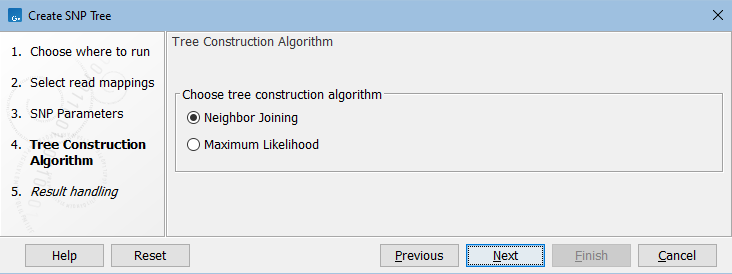
Figure 9.3: Choose the tree construction algorithm.
- Neighbor Joining. Branch length is based on the distance between samples computed as Positions used where the consensus sequence is different / Total positions. If you move from one sample to another in the tree, the sum of the lengths of the branches traversed equals the distance between those samples.
- Maximum Likelihood. Creates an initial tree using the Neighbor Joining method and subsequently calculates the most likely phylogenetic tree under the given evolutionary model. Parameters for this method are defined in the next dialog (see figure 9.4).
If you selected Maximum Likelihood, the next dialog covers parameters for this algorithm (see figure 9.4). The parameters are described here: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Maximum_Likelihood_Phylogeny.html.
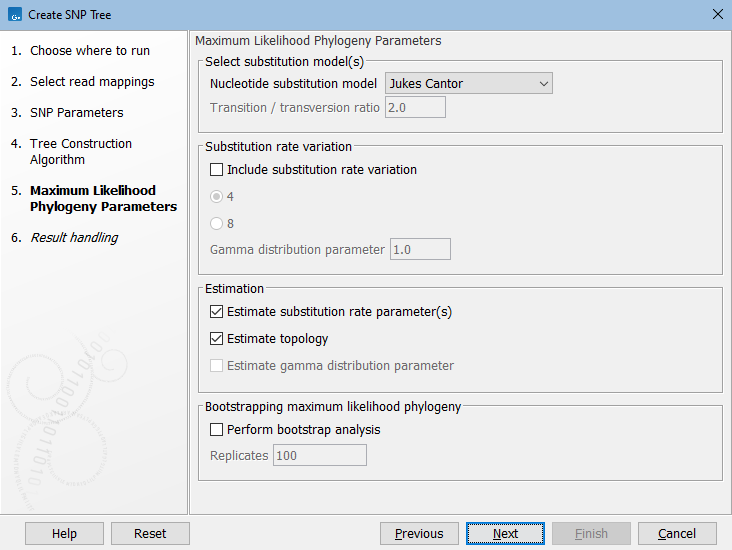
Figure 9.4: Set parameters for maximum likeihood estimation.
In the Result handling dialog, specify the output (figure 9.5).
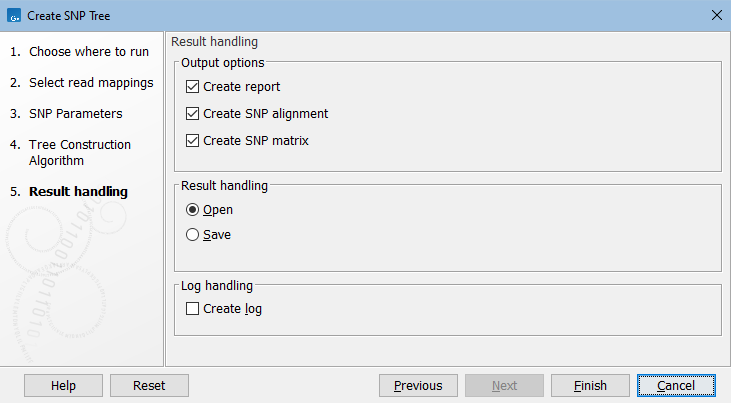
Figure 9.5: Create SNP Tree output options.
In addition to the SNP tree, the following are available:
- Create report. A SNP report with summary of input and results.
- Create SNP alignment. Outputs the alignment of concatenated SNPs that is produced as a first step in the algorithm.
The alignment can be used as input for the Model Testing tool that serves to identify which evolutionary model suits the data best. Based on this, you may want to rerun the Create SNP Tree tool with adjusted settings. The Model Testing tool is described at https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Model_Testing.html.
- Create SNP matrix. A matrix containing the number of SNP differences between all pairs of samples.
Subsections