Optional Merge Paired Reads
The Optional Merge Paired Reads tool has been moved to Legacy Tools folder because it is now incorporated into the OTU Clustering tool. When the input consists of paired reads, the OTU clustering tool will initially merge them into pairs, and align the resulting paired reads to OTUs. Reads that cannot be merged will be independently aligned to reference OTUs. Then both reads of a pair will be assigned to the OTU where they both align with the highest identity possible. Finally, the tool merges both reads of the pair using a stretch of N to the fragments so that the paired read looks as much as possible like the OTU they have been assigned to. For example, the forward-reverse pair (ACGACGACG, GTAGTAGTA) will be turned into ACGACGACGnnnnnnnnnnnnnnnnnnnnTACTACTAC.
In order to use the highest quality sequences for clustering, it is recommended to merge paired read data. If the read length is smaller than the amplicon size, forward and reverse reads are expected to overlap in most of their 3' regions. Therefore, one can merge the forward and reverse reads to yield one high quality representative using the Optional Merge Paired Reads tool. The Optional Merge Paired Reads tool will merge paired-end reads according to some pre-selected merge parameters: the overlap region and the quality of the sequences. For example, for a designed 150 bp overlap, a maximum score of 150 is achievable, but as the real length of the overlap is unknown, a lower minimum score should be chosen. Also, some mismatches and indels should be allowed, especially if the sequence quality is not perfect. You can also set penalties for mismatch, gap and unaligned ends.
To run the Optional Merge Paired Reads tool, go to Microbial Genomics Module (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Amplicon-based OTU clustering (
) | Amplicon-based OTU clustering (![]() ) | Optional Merge Paired Reads (
) | Optional Merge Paired Reads (![]() ).
).
Select any number of sequences as input. The tool accepts both paired and unpaired reads but will only merge the paired reads while returning the unpaired ones as "not merged" reads in the output. Note that paired reads have to be in forward-reverse orientation. After merging, the merged reads will always be in the forward orientation.
Click Next to open the dialog shown in figure 16.1.
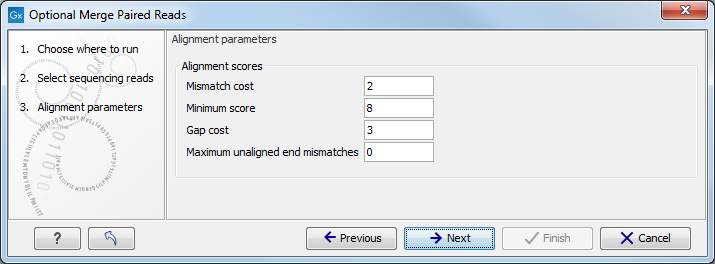
Figure 16.1:
Alignment parameters.
In order to understand how these parameters should be set, an explanation of the merging algorithm is needed: Because the fragment size is not an exact number of base pairs and is different from fragment to fragment, an alignment of the two reads has to be performed. If the alignment is good and long enough, the reads will be merged. Good enough in this context means that the alignment has to satisfy some user-specified score criteria (details below). Because of sequencing errors that typically are more abundant towards the end of the read, the alignment is not expected always to be perfect, and the user can decide how many errors are acceptable. Long enough in this context means that the overlap between the reads has to be non-coincidental. Merging two reads that do not really overlap leads to errors in the downstream analysis, thus it is very important to make sure that the overlap is big enough. If only a few bases overlap was required, some read pairs will match by chance, so this has to be avoided.
The following parameters are used to define what is good enough and long enough.
- Mismatch cost: The alignment awards one point for a match, and the mismatch cost is set by this parameter. The default value is 2.
- Minimum score: This is the minimum score of an alignment to be accepted for merging. The default value is 8. As an example: with default settings, this means that an overlap of 11 bases with one mismatch will be accepted (10 matches minus 2 for a mismatch).
- Gap cost: This is the cost for introducing an insertion or deletion in the alignment. The default value is 3.
- Maximum unaligned end mismatches: The alignment is local, which means that a number of bases can be left unaligned. If the quality of the reads is dropping to be very poor towards the end of the read, and the expected overlap is long enough, it makes sense to allow some unaligned bases at the end. However, this should be used with great care which is why the default value is 0. As explained above, a wrong decision to merge the reads leads to errors in the downstream analysis, so it is better to be conservative and accept fewer merged reads in the result. Please note that even with the alignment scores above the minimum score specified in the tool setup, the paired reads also need to have the number of end mismatches below the "Maximum unaligned end mismatches" value specified in the tool setup to be qualified for merging.
The main result will be two sequence lists for each sample selected as input to the tool: one containing the merged reads (labeled as "merged"), and one containing the reads that could not be merged (labeled as "not merged"). Note that low quality can be one of the reasons why a pair cannot be merged. Hence, the list of reads that could not be paired is more likely to contain more reads with errors than the one with the merged reads.