Create SNP Tree
The Create SNP Tree tool is inspired by [Kaas et al., 2014]. There are two ways to initiate creation of a SNP tree: from the Result Metadata Table (see subsection 9.4) or by running the tool from the Toolbox. Note that you can only create a SNP tree if you have identified a common reference for the different stains you are trying to type, and used it for read mapping and variant calling for each of these samples.
To create a SNP tree from the Toolbox:
Microbial Genomics Module (![]() ) | Typing and Epidemiology (beta) (
) | Typing and Epidemiology (beta) (![]() ) | Create SNP Tree (
) | Create SNP Tree (![]() )
)
Select the relevant read mappings as shown in figure 13.1
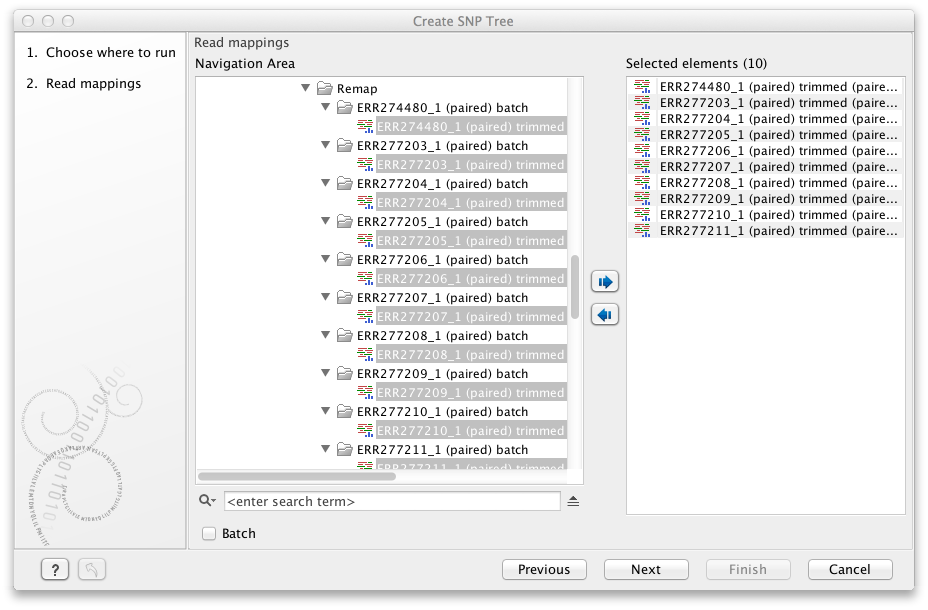
Figure 13.1: Select read mappings to be included in the SNP tree analysis.
Alternatively, select data recursively by right-clicking on the folder name and selecting Add folder contents (recursively) (figure 13.2), but remember to double check that files relevant for the downstream analysis are selected. An efficient alternative to these methods is to use the Quick filtering functionality from the Metadata Result Table to filter easily the data and initiate the SNP tree creation.
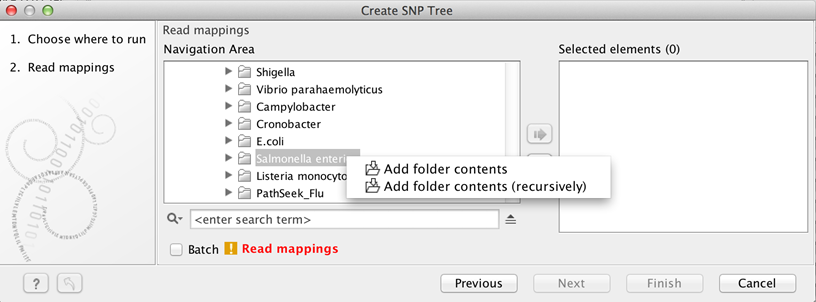
Figure 13.3: For selection of all sequence files in a folder, right click and select Add folder contents (recursively).
Select the variant tracks you want to use (figure 13.3). The variant tracks determine which positions to include in the SNP tree. The variant tracks need to have the same reference as the previously selected read mappings. Under normal circumstances you would select one variant track for each read mapping given in the input step, but that is not a requirement.
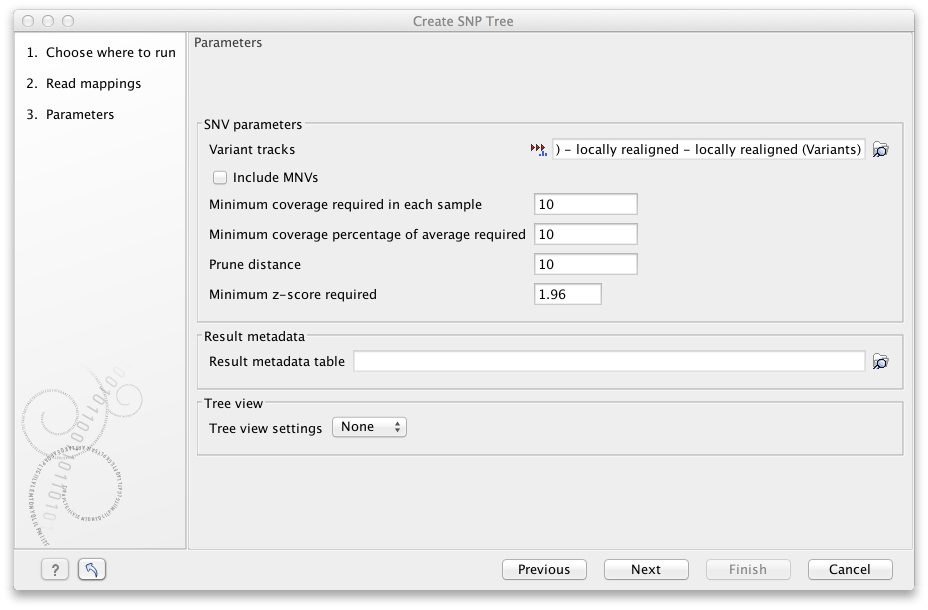
Figure 13.2: Select variant tracks and specify relevant parameters before generation of a SNP tree.
The following Parameters may be specified before the generation of the SNP tree (see figure 13.3):
- SNV parameters
- Variant tracks. Select the variant tracks you want to use. The variant tracks determine which positions to include in the SNP tree.
- Include MNVs or not, along with SNVs when building the SNP tree.
- Minimum coverage required in each sample on a given position. The position is skipped if at least one sample has coverage below the specified threshold.
- Minimum coverage percentage of average required on a given position. The position is skipped if at least one sample has coverage below this percentage of its own average coverage.
- Prune distance specifies the minimum number of nucleotides between unfiltered positions. If a position is within this distance of a previously used position it will be filtered.
- Minimum z-score required. Defining
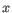 as the number of most prevalent nucleotide at a position and
as the number of most prevalent nucleotide at a position and 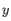 as the coverage subtracting , the z-score is calculated as
as the coverage subtracting , the z-score is calculated as
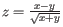 . If the calculated z-score for a given position is less than the specified minimum value the position is filtered.
. If the calculated z-score for a given position is less than the specified minimum value the position is filtered.
- Result metadata
- Result metadata Table. Specify location of the Result metadata table file.
- Tree view
- Tree view settings. Select a standard tree setting (i.e., None, K-mer Tree Default or SNP Tree Default) or your own custom tree setting. Read more on Tree Settings in general: http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Tree_Settings.html.
The variant calls and read mapping results are used to determine the SNP positions used in the tree. Note that the variant tracks are only used to determine which positions to include in the SNP tree. Only the position and the type (SNP, and MNV if enabled) are used, whereas any information about reference and allele is ignored. The read mappings are then used to estimate the consensus sequence. Only a variant with relative frequency above 50% (haploid organisms) will be effectively considered.
The initial list of variants is reduced as the following: All but one variant from the initial variant lists that fall within the specified pruning distance (for example 10nt) are ignored. Positions that are not well or not covered in one or more read mappings ("Minimum coverage required in each sample" and "Minimum coverage of average required") are removed. In addition, all SNPs which do not have the minimal z-score are excluded.
The Neighbour Joining method is used to create the tree. Branch lengths are based on the distance between samples. The distance between two samples is computed as "Number of input positions used where the consensus sequence is different" / "Number of input positions used". The distance is therefore a number between 0 (no difference found in the input positions used) and 1 (all input positions used were different). From the tree, one can compute the distance between two samples by summing up all branches connecting them.
Subsections
- SNP tree output report
- Visualization of SNP Tree including metadata and analysis result metadata
- SNP Tree Variants editor viewer