De Novo Assemble Metagenome
Adapters should be removed from sequences before assembly using the Trim Sequences tool. The presence of adapters can result in the assembler trying to join regions that are not biologically relevant, leading to an assembly taking a long time and yielding misleading results. Quality trimming before assembling is not generally necessary as the assembler itself should weed out or correct bad quality regions. However, trimming off low quality regions may decrease the amount of memory needed for the de novo assembly, which can be an advantage when working with large datasets.
To assemble a metagenome de novo, run the tool:
Microbial Genomics Module (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Functional Analysis (
) | Functional Analysis (![]() ) | De Novo Assemble Metagenome (
) | De Novo Assemble Metagenome (![]() )
)
Select one or more sequence lists or single sequences to assemble, then set the parameters for the assembly. This will show a dialog similar to the one in figure 6.1.
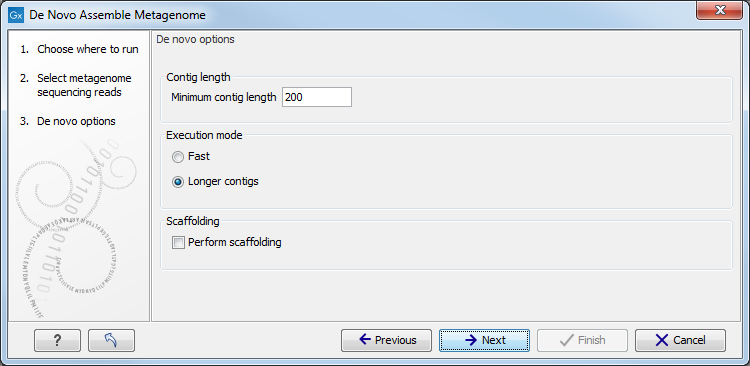
Figure 6.1: Setting parameters for the assembly.
The first parameter allows you to specify the Minimum contig length. Contigs below this length (default value 200 bp) will not be reported. The assembler will often produce shorter contigs for very complex datasets containing reads from many closely related species. In such a case, it is often wise to set a lower threshold in order to cover a larger proportion of the metagenome with contigs. Similarly, for metagenomes of low complexity, it is often wise to set a higher threshold in order to avoid duplication.
After setting the Minimum contig length, you need to choose between running the assembler in Fast mode or Longer contigs mode. In Fast mode, the assembler is iterated once with a predifined wordsize (![]() ). In Longer contigs mode, the assembler is iterated three times with increasing wordsize (
). In Longer contigs mode, the assembler is iterated three times with increasing wordsize (
![]() ), using the contigs from the previous iteration as input in the next iteration together with the input reads. Fast mode produces contigs of very high quality very fast, while the Longer contigs mode produces significantly longer contigs (possibly with slightly more misassemblies). Longer contigs mode naturally requires up to three times more compute time.
), using the contigs from the previous iteration as input in the next iteration together with the input reads. Fast mode produces contigs of very high quality very fast, while the Longer contigs mode produces significantly longer contigs (possibly with slightly more misassemblies). Longer contigs mode naturally requires up to three times more compute time.
At the bottom, there is an option to Perform scaffolding. When this option is selected, the assembler will attempt to join contigs using paired-end information as the last step of the assembly process. Since paired-end information is needed to perform scaffolding, this option is disabled if the input dataset does not contain any paired-end reads.
The result of the de novo assembly is a list of contigs in a sequence list. If the Perform scaffolding option was selected in the previous wizard step, the resulting scaffolds will appear at the end of the sequence list and will be named scaffold_1, scaffold_2, etc. If Create report was selected, the tool will create a summary report containing basic statistics on both the input reads and the output contigs.