Motif search from the Toolbox
The dynamic motifs provide a quick way of routinely scanning a sequence for commonly used motifs, but in some cases a more systematic approach is needed. The motif search in the Toolbox provides an option to search for motifs with a user-specified similarity to the target sequence, and furthermore the motifs found can be displayed in an overview table. This is particularly useful when searching for motifs on many sequences.
To start the Toolbox motif search, go to:
Toolbox | Classical Sequence Analysis (![]() ) | General Sequence Analysis (
) | General Sequence Analysis (![]() )| Motif Search
(
)| Motif Search
(![]() )
)
A dialog window will be launched. Use the arrows to add or remove sequences or sequence lists between the Navigation Area and the selected elements.
You can perform the analysis on several DNA or several protein sequences at a time. In this case, the method will search for patterns in the sequences and create an overview table of the motifs found in all sequences.
Click Next to adjust parameters (see figure 14.30).
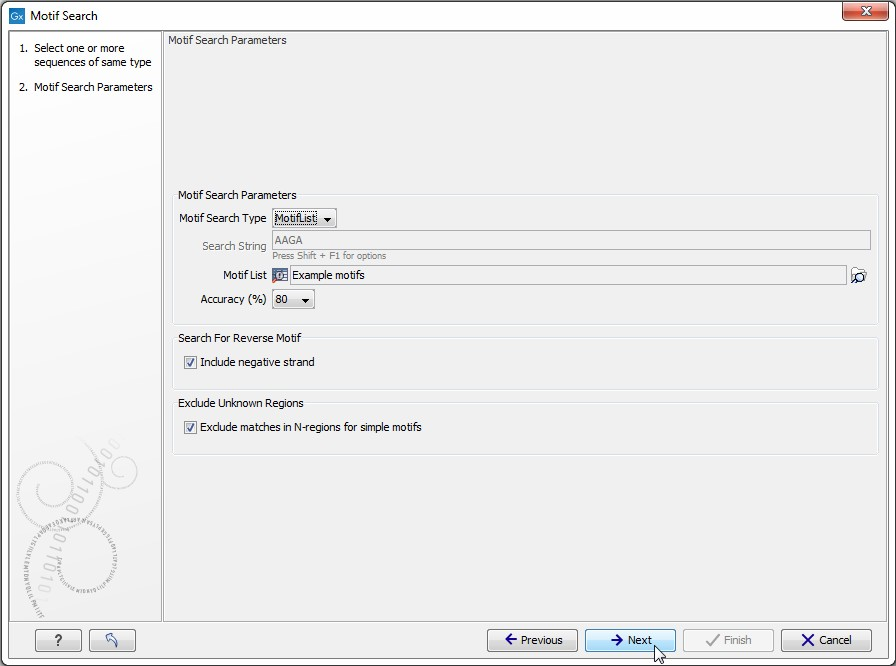
Figure 14.30: Setting parameters for the motif search.
The options for the motif search are:
- Motif types. Choose what kind of motif to be used:
- Simple motif. Choosing this option means that you enter a simple motif, e.g. ATGATGNNATG.
- Java regular expression. See Java regular expressions.
- Prosite regular expression. For proteins, you can enter different protein patterns from the PROSITE database (protein patterns using regular expressions and describing specific amino acid sequences). The PROSITE database contains a great number of patterns and have been used to identify related proteins (see http://www.expasy.org/cgi-bin/prosite-list.pl).
- Use motif list. Clicking the small button (
 ) will allow you to select a saved motif list (see Create motif lists).
) will allow you to select a saved motif list (see Create motif lists).
- Motif. If you choose to search with a simple motif, you should enter a literal string as your motif. Ambiguous amino acids and nucleotides are allowed. Example; ATGATGNNATG. If your motif type is Java regular expression, you should enter a regular expression according to the syntax rules described here. Press Shift + F1 key for options. For proteins, you can search with a Prosite regular expression and you should enter a protein pattern from the PROSITE database.
- Accuracy. If you search with a simple motif, you can adjust the accuracy of the motif to the match on the sequence. If you type in a simple motif and let the accuracy be 80%, the motif search algorithm runs through the input sequence and finds all subsequences of the same length as the simple motif such that the fraction of identity between the subsequence and the simple motif is at least 80%. A motif match is added to the sequence as an annotation with the exact fraction of identity between the subsequence and the simple motif. If you use a list of motifs, the accuracy applies only to the simple motifs in the list.
- Search for reverse motif. This enables searching on the negative strand on nucleotide sequences.
- Exclude unknown regions. Genome sequence often have large regions with unknown sequence. These regions are very often padded with N's. Ticking this checkbox will not display hits found in N-regions.Motif search handles ambiguous characters in the way that two residues are different if they do not have any residues in common. For example: For nucleotides, N matches any character and R matches A,G. For proteins, X matches any character and Z matches E,Q.
- Add annotations. This will add an annotation to the sequence when a motif is found (an example is shown in figure 14.31.
- Create table. This will create an overview table of all the motifs found for all the input sequences.
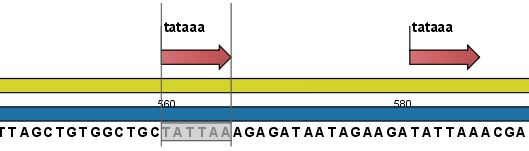
Figure 14.31: Sequence view displaying the pattern found. The search string was 'tataaa'.