The phylogenetic tree
The evolutionary hypothesis of a phylogeny can be graphically represented by a phylogenetic tree.
Figure 22.10 shows a proposed phylogeny for the great apes, Hominidae, taken in part from Purvis [Purvis, 1995]. The tree consists of a number of nodes (also termed vertices) and branches (also termed edges). These nodes can represent either an individual, a species, or a higher grouping and are thus broadly termed taxonomic units. In this case, the terminal nodes (also called leaves or tips of the tree) represent extant species of Hominidae and are the operational taxonomic units (OTUs). The internal nodes, which here represent extinct common ancestors of the great apes, are termed hypothetical taxonomic units since they are not directly observable.
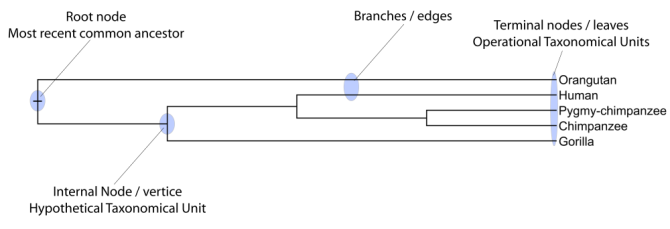
Figure 22.10: A proposed phylogeny of the great apes (Hominidae). Different components of the tree are marked, see text for description.
The ordering of the nodes determine the tree topology and describes how lineages have diverged over the course of evolution. The branches of the tree represent the amount of evolutionary divergence between two nodes in the tree and can be based on different measurements. A tree is completely specified by its topology and the set of all edge lengths.
The phylogenetic tree in figure 22.10 is rooted at the most recent common ancestor of all Hominidae species, and therefore represents a hypothesis of the direction of evolution e.g. that the common ancestor of gorilla, chimpanzee and man existed before the common ancestor of chimpanzee and man. In contrast, an unrooted tree would represent relationships without assumptions about ancestry.
Besides evolutionary biology and systematics the inference of phylogenies is central to other areas of research.
As more and more genetic diversity is being revealed through the completion of multiple genomes, an active area of research within bioinformatics is the development of comparative machine learning algorithms that can simultaneously process data from multiple species [Siepel and Haussler, 2004]. Through the comparative approach, valuable evolutionary information can be obtained about which amino acid substitutions are functionally tolerant to the organism and which are not. This information can be used to identify substitutions that affect protein function and stability, and is of major importance to the study of proteins [Knudsen and Miyamoto, 2001]. Knowledge of the underlying phylogeny is, however, paramount to comparative methods of inference as the phylogeny describes the underlying correlation from shared history that exists between data from different species.
In molecular epidemiology of infectious diseases, phylogenetic inference is also an important tool. The very fast substitution rate of microorganisms, especially the RNA viruses, means that these show substantial genetic divergence over the time-scale of months and years. Therefore, the phylogenetic relationship between the pathogens from individuals in an epidemic can be resolved and contribute valuable epidemiological information about transmission chains and epidemiologically significant events [Leitner and Albert, 1999], [Forsberg et al., 2001].