Adapter trimming
Clicking Next will allow you to specify parameters for adapter trimming.
When you are analyzing sequencing data, the adapters must be trimmed off before you proceed with further analysis. The removal of adapters is often done directly on the sequencing machine, but in some cases, some adapters remain on the sequenced reads. The presence of remaining adapters can lead to misleading results, so we recommend to trim them off the reads (figure 25.3).
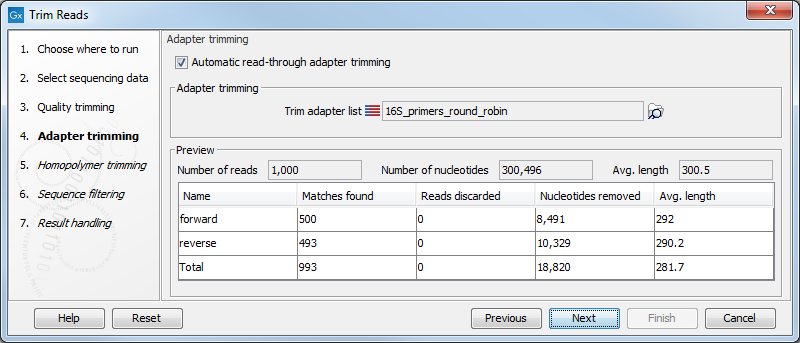
Figure 25.3: Trimming your sequencing data for adapter sequences.
The default option for this trimming step is to use the "Automatic read-through adapter trimming", which will detect read-through adapter sequence on paired-end reads automatically. Read-through means that the sample DNA fragment being sequenced is shorter than the read length, such that the 3' end of one read includes the reverse-complement of the adapter from the start of the other read. Leaving this option enabled is always recommended: the trimming performed automatically can detect read-through of even a single nucleotide, which is not the case when trimming using a trim adapter list. The detected adapters for the first and second read can be found in the Trim Reads report.
There are however a couple of limitations on the "Automatic read-through adapter trimming" option: this option detects overlap in paired reads containing standard nucleotides (A, T, C, and G). If the read contains ambiguous symbols, such as N, these will not match the standard nucleotides.
Also, the first and second read should be of equal (or near-equal) length - some sequencing protocols use asymmetric read lengths for the first and second read, in which case the tool is less likely to detect and trim the read-through.
So when you are working with data of low quality, asymmetric read lengths, mate-paired reads, single reads, small RNAs, or also when working with gene specific primers, it is recommended that you specify a trim adapter read in addition to using the "Automatic read-through adapter trimming" option. It is even possible to use the report of the Trim Read tool to find out what Trim adapter list should be used for the data at hand. Read Trim adapter list to learn how to create an adapter list.
Below you find a preview listing the results of trimming with the adapter trimming list on 1000 reads in the input file (reads 1001-2000 when the read file is long enough). This is useful for a quick feedback on how changes in the parameters affect the trimming (rather than having to run the full analysis several times to identify a good parameter set). The following information is shown:
- Name. The name of the adapter.
- Matches found. Number of matches found based on the settings.
- Reads discarded. This is the number of reads that will be completely discarded. This can either be because they are completely trimmed (when the Action is set to Remove adapter and the match is found at the 3' end of the read), or when the Action is set to Discard when found or Discard when not found.
- Nucleotides removed. The number of nucleotides that are trimmed include both the ones coming from the reads that are discarded and the ones coming from the parts of the reads that are trimmed off.
- Avg. length This is the average length of the reads that are retained (excluding the ones that are discarded).