BLAST against local data
Running BLAST searches on your local machine can have several advantages over running the searches remotely at the NCBI:
- It can be faster.
- It does not rely on having a stable internet connection.
- It does not depend on the availability of the NCBI BLAST servers.
- You can use longer query sequences.
- You use your own data sets to search against.
There are a number of options for what you can search against:
- You can create a database based on data already imported into your Workbench (see Create BLAST database)
- You can add pre-formatted databases (see Download BLAST databases)
- You can use sequence data from the Navigation Area directly, without creating a database first.
To conduct a local BLAST search, go to:
Toolbox | BLAST (![]() )| BLAST (
)| BLAST (![]() )
)
This opens the dialog seen in figure 13.5:
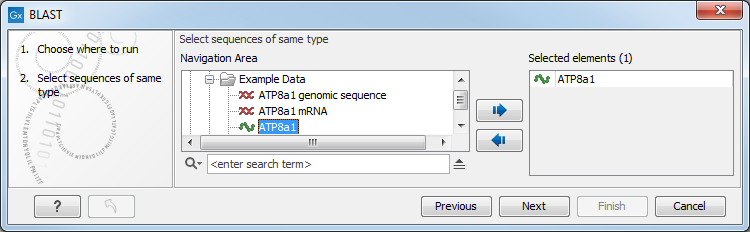
Figure 13.5: Choose one or more sequences to conduct a BLAST search.
Select one or more sequences of the same type (DNA or protein) and click Next.
This opens the dialog seen in figure 13.6:
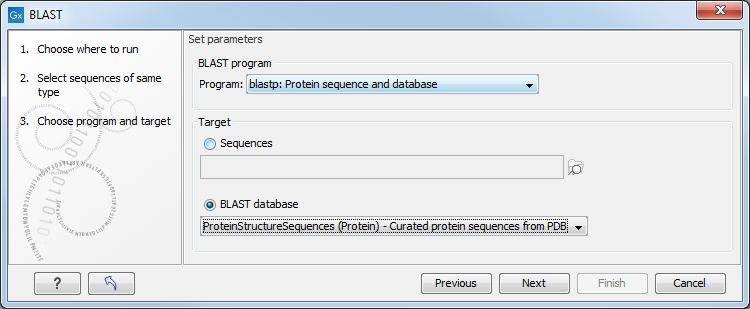
Figure 13.6: Choose a BLAST program and a target database.
At the top, you can choose between different BLAST programs.
BLAST programs for DNA query sequences:
- blastn: DNA sequence against a DNA database. Searches for DNA sequences with homologous regions to your nucleotide query sequence.
- blastx: Translated DNA sequence against a Protein database. Automatic translation of your DNA query sequence in six frames; these translated sequences are then used to search a protein database.
- tblastx: Translated DNA sequence against a Translated DNA database. Automatic translation of your DNA query sequence and the DNA database, in six frames. The resulting peptide query sequences are used to search the resulting peptide database. Note that this type of search is computationally intensive.
BLAST programs for protein query sequences:
- blastp: Protein sequence against Protein database. Used to look for peptide sequences with homologous regions to your peptide query sequence.
- tblastn: Protein sequence against Translated DNA database. Peptide query sequences are searched against an automatically translated, in six frames, DNA database.
In cases where you have selected blastx or tblastx to conduct a search, you will get the option of selecting a translation table for the genetic code. The standard genetic code is set as default. This setting is particularly useful when working with organisms or organelles that have a genetic code that differs from the standard genetic code.
If you search against the Protein Data Bank database and homologous sequences are found to the query sequence, these can be downloaded and opened with the 3D Molecule Viewer (see BLAST search against the PDB database).
You then specify the target database to use:
- Sequences. When you choose this option, you can use sequence data from the Navigation Area as database by clicking the Browse and select icon (
 ). A temporary BLAST database will be created from these sequences and used for the BLAST search. It is deleted afterwards. If you want to be able to click in the BLAST result to retrieve the hit sequences from the BLAST database at a later point, you should not use this option; create a create a BLAST database first.
). A temporary BLAST database will be created from these sequences and used for the BLAST search. It is deleted afterwards. If you want to be able to click in the BLAST result to retrieve the hit sequences from the BLAST database at a later point, you should not use this option; create a create a BLAST database first.
- BLAST Database. Select a database already available in one of your designated BLAST database folders. Read more in Manage BLAST databases.
When a database or a set of sequences has been selected, click Next.
The next dialog allows you to adjust the parameters to meet the requirements of your BLAST search (figure 13.7).
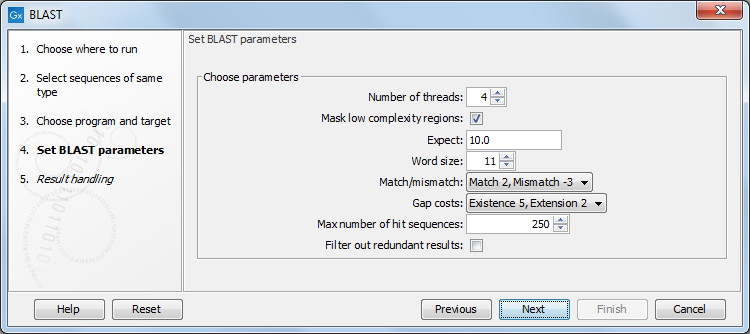
Figure 13.7: Parameters that can be set before submitting a local BLAST search.
- Number of threads. You can specify the number of threads, which should be used if your Workbench is installed on a multi-threaded system.
- Mask low complexity regions. Mask off segments of the query sequence that have low compositional complexity. Filtering can eliminate statistically significant, but biologically uninteresting reports from the BLAST output (e.g. hits against common acidic-, basic- or proline-rich regions), leaving the more biologically interesting regions of the query sequence available for specific matching against database sequences.
- Expect. The threshold for reporting matches against database sequences. The Expect value (E-value) describes the number of hits one can expect to see matching a query by chance when searching against a database of a given size. If the E-value ascribed to a match is greater than the value entered in the Expect field, the match will not be reported. Details of how E-values are calculated can be found at the NCBI: http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html. Lower thresholds are more stringent, leading to fewer chance matches being reported. Increasing the threshold results in more matches being reported, but many may just matching by chance, not due to any biological similarity. Values lower than 1 can be entered as decimals, or in scientific notiation. For example, 0.001, 1e-3 and 10e-4 would be equivalent and acceptable values.
- Word Size. BLAST is a heuristic that works by finding word-matches between the query and database sequences. You may think of this process as finding "hot-spots" that BLAST can then use to initiate extensions that might lead to full-blown alignments. For nucleotide-nucleotide searches (i.e. "BLASTn") an exact match of the entire word is required before an extension is initiated, so that you normally regulate the sensitivity and speed of the search by increasing or decreasing the wordsize. For other BLAST searches non-exact word matches are taken into account based upon the similarity between words. The amount of similarity can be varied so that you normally uses just the wordsizes 2 and 3 for these searches.
- Match/mismatch. A key element in evaluating the quality of a pairwise sequence alignment is the "substitution matrix", which assigns a score for aligning any possible pair of residues. The matrix used in a BLAST search can be changed depending on the type of sequences you are searching with (see the BLAST Frequently Asked Questions). Only applicable for protein sequences or translated DNA sequences.
- Gap Cost. The pull down menu shows the Gap Costs (Penalty to open Gap and penalty to extend Gap). Increasing the Gap Costs and Lambda ratio will result in alignments which decrease the number of Gaps introduced.
- Max number of hit sequences. The maximum number of database sequences, where BLAST found matches to your query sequence, to be included in the BLAST report.
- Filter out redundant results. This option culls HSPs on a per subject sequence basis by removing HSPs that are completely enveloped by another HSP.
BLAST a partial sequence against a local database
You can search a database using only a part of a sequence directly from the sequence view:
select the region that you wish to BLAST |
right-click the selection | BLAST Selection Against
Local Database (![]() )
)
This will go directly to the dialog shown in figure 13.6 and the rest of the options are the
same as when performing a BLAST search with a full sequence.