Perform QIAseq Immune Repertoire Analysis ready-to-use workflow
Using RNA sequencing data as input the Perform QIAseq Immune Repertoire Analysis workflow can be used to characterize the T cell receptor repertoire. The workflow includes all necessary steps for processing the RNA sequencing reads such as UMI grouping, trimming off the constant (C) regions of the read and characterizing the T cell receptor repertoire.
|
Due to licensing considerations, the reference data required to run this workflow could not be made available at the time of the Biomedical Genomics Analysis 20.1 release. To check if the reference data is available when you plan to run this workflow, please open the Reference Data Manager by clicking on the References button in the top toolbar. Click on the QIAGEN Sets tab and look for these reference sets:
|
To run the Perform QIAseq Immune Repertoire Analysis workflow, go to the toolbox and find:
Ready-to-Use Workflows | QIAseq Panel Analysis (![]() ) | QIAseq Analysis Workflows (
) | QIAseq Analysis Workflows (![]() ) | Perform QIAseq Immune Repertoire Analysis (
) | Perform QIAseq Immune Repertoire Analysis (![]() )
)
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible. Click on the button labeled Next.
Select the RNA sequencing reads to analyze by locating them in the Navigation Area and either double-click on the sample name or use the arrow pointing to the right to select the RNA reads. When analyzing more than one sample at the time, the Batch checkbox in the lower left corner of the dialog should be checked. Click on the button labeled Next.
In this step you can specify the relevant Reference Data Set. Reference data sets for human and mouse are provided by QIAGEN. Each reference data set consists of an adapter trim list used for trimming off the constant region of the T cell receptor sequences as well as reference sequences for the TCR V- and J genes. In the left-hand side the available reference data sets are listed:
- QIAseq Immune Repertoire Analysis IMGT Reference Sequences for analysis of human data
- QIAseq Immune Repertoire Analysis Mouse IMGT Reference Sequences for analysis of mouse data
If you have not downloaded the Reference Data Set yet, the dialog will suggest the relevant data set and offer the opportunity to download the reference data to where you have chosen to run the workflow; Download to Workbench or Download to Server. In the example shown in figure 7.1 the check mark to the left of the human reference data indicates that this reference data set has already been downloaded to the server and is ready to use. The plus symbol next to the mouse reference data indicates that this reference data set has not yet been downloaded. To download the mouse data to the server, click on the text QIAseq Immune Repertoire Analysis Mouse IMGT Reference Sequences to enable the button labeled Download to Server.
If the Reference Data Set was previously downloaded, the option "Use the default reference data" is available and will ensure the relevant reference data set is used. You can always check the "Select a reference data set to use" option to be able to specify another Reference Data Set than the one suggested.
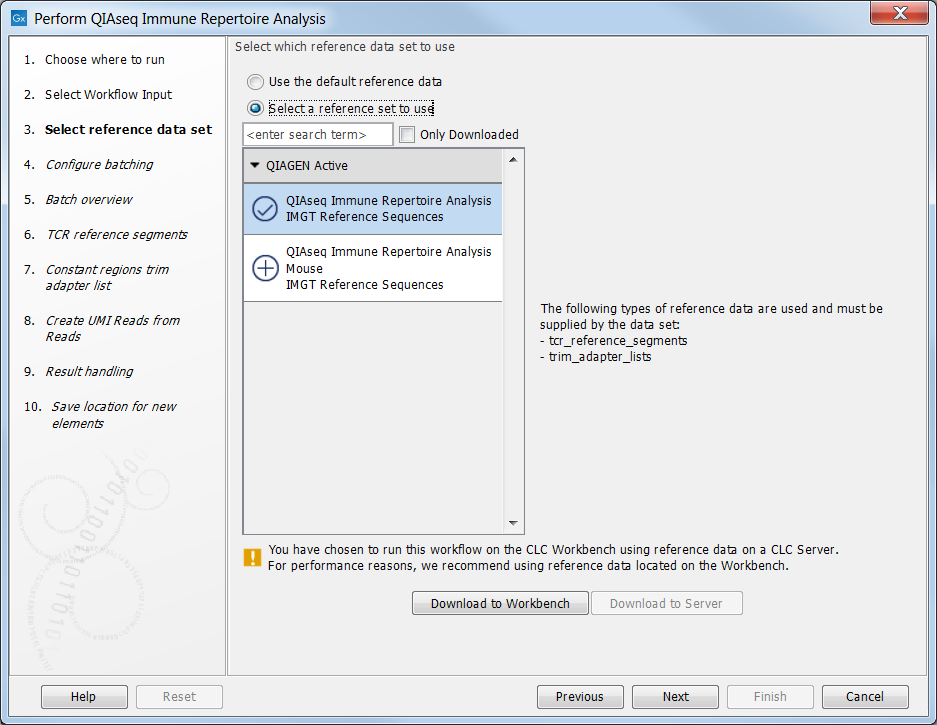
Figure 7.1: The Reference Data Set for analysis of human data is highlighted. The references needed by the workflow are listed to the right and to the left, you can find a list of reference data sets available for this specific ready-to-use workflow. For this workflow reference data are available for human and mouse data.
When running the workflow in Batch mode an extra wizard step will appear where you get the option to associate metadata to the analysis. More information about metadata usage can be found here http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Metadata.html.
If the batch option was checked, a dialog will now appear where the batch units should be defined. Choose Use organization of input data if you want to run the analysis without metadata usage. Choose Use metadata if you would like to associate metadata to the analysis. To associate metadata, you must select a metadata table. Information about how to import or create a metadata table can be found here http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Creating_metadata_tables.html.
If the analysis is run in batch-mode the next dialog will show an overview of the samples, otherwise clicking on the button labeled Next will bring up a dialog where you can see the selected TCR reference sequence data. Click on the folder icon on the right-hand side if you want to select other TCR reference sequence data. Click Next to go to the next step where you can check the selected adapter trim list for trimming off the constant regions.
Click on the button labeled Next to specify the Minimum UMI group size. This number specifies the minimum number of UMI reads that is required before a UMI group is being considered. Reads belonging to UMI groups supported by fewer reads than this number will be discarded. More information about UMI groups can be found here Unique Molecular Index Groups and Create UMI Reads from Reads
Finally, choose where to save the data, and press Finish to start the analysis.
Subsections