Identify and Annotate Variants (WES-HD)
The Identify and Annotate Variants (WES-HD) tool should be used to identify and annotate variants in one sample. The tool consists of a workflow that is a combination of the Identify Variants and the Annotate Variants workflows.
The tool runs an internal workflow, which starts with mapping the sequencing reads to the human reference sequence. Then it runs a local realignment to improve the variant detection, which is run afterwards. After the variants have been detected, they are annotated with gene names, amino acid changes, conservation scores, information from relevant variants present in the ClinVar database, and information from common variants present in the common dbSNP Common, HapMap, and 1000 Genomes database. Furthermore, a targeted region report is created to inspect the overall coverage and mapping specificity.
The difference between Identify and Annotate Variants (TAS-HD) and (WES-HD) is that the Autodetect paired distances has been switched off in Map Reads to Reference tool for the TAS workflows.
Run the Identify and Annotate Variants (WES-HD) workflow
To run the Identify and Annotate Variants (WES-HD) workflow, go to:
Toolbox | Ready-to-Use Workflows | Whole Exome Sequencing (![]() ) | Hereditary Disease (
) | Hereditary Disease (![]() ) | Identify and Annotate Variants (WES-HD) (
) | Identify and Annotate Variants (WES-HD) (![]() )
)
- Double-click on the Identify and Annotate Variants (WES-HD) tool to start the analysis. If you are connected to a server, you will first be asked where you would like to run the analysis.
- Select the sequencing reads you want to analyze (figure 19.82).
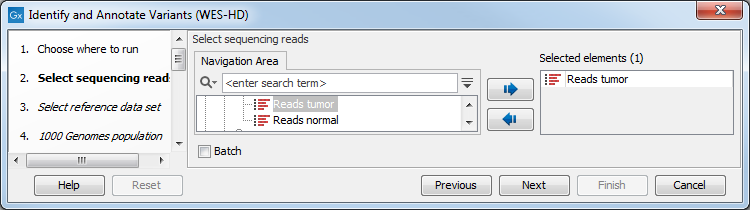
Figure 19.82: Specify the sequencing reads for the appropriate family member. - In the next dialog, you have to select which data set should be used to identify variants (figure 19.83).
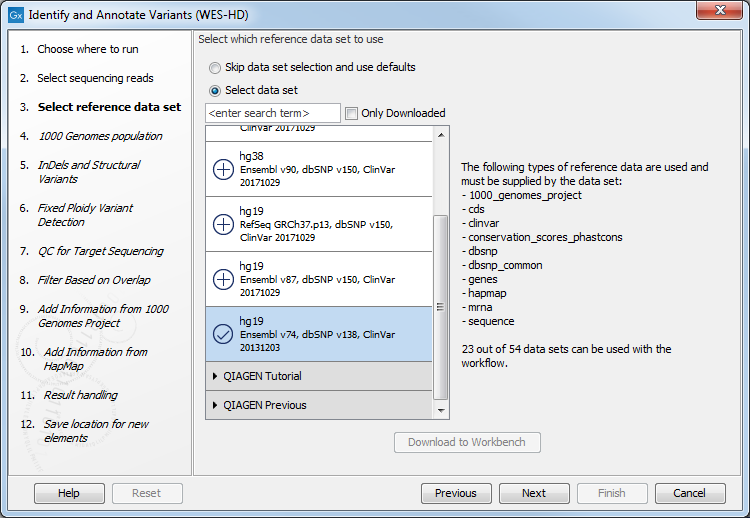
Figure 19.83: Choose the relevant reference Data Set to identify variants. - Specify which 1000 Genomes population you would like to use (figure 19.84).
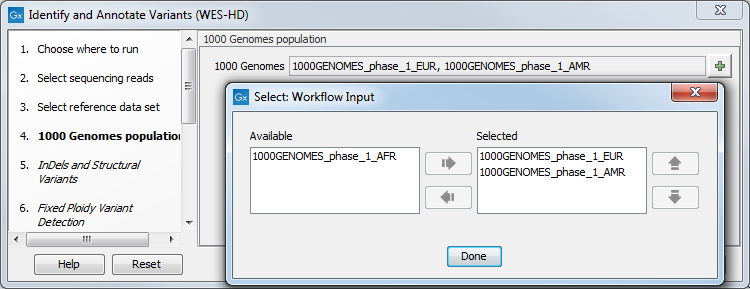
Figure 19.84: Select the relevant 1000 Genomes population(s). - Specify a target region file for the Indels and Structural Variants tool.
(figure 19.85).
The targeted region file is a file that specifies which regions have been sequenced, when working with whole exome sequencing or targeted amplicon sequencing data. This file is something that you must provide yourself, as this file depends on the technology used for sequencing. You can obtain the targeted regions file from the vendor of your targeted sequencing reagents.
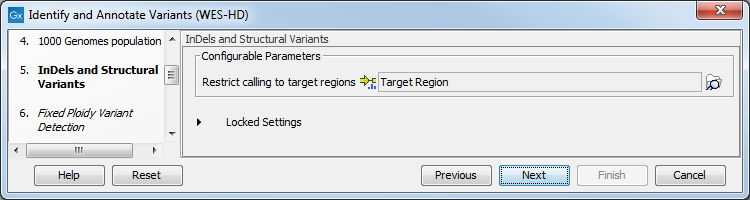
Figure 19.85: Specify the parameters for the Indels and Structural Variants tool. - Specify the Fixed Ploidy Variant Detection settings, including a target region file (figure 19.86).
The parameters used by the Fixed Ploidy Variant Detection tool can be adjusted. We have optimized the parameters to the individual analyses, but you may want to tweak some of the parameters to fit your particular sequencing data. A good starting point could be to run an analysis with the default settings.
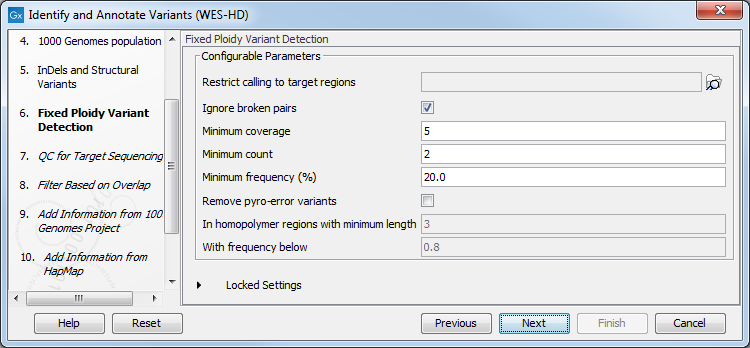
Figure 19.86: Specify the parameters for the Fixed Ploidy Variant Detection tool.The parameters that can be set are:
- Required variant probability is the minimum probability value of the 'variant site' required for the variant to be called. Note that it is not the minimum value of the probability of the individual variant. For the Fixed Ploidy Variant detector, if a variant site - and not the variant itself - passes the variant probability threshold, then the variant with the highest probability at that site will be reported even if the probability of that particular variant might be less than the threshold. For example if the required variant probability is set to 0.9 then the individual probability of the variant called might be less than 0.9 as long as the probability of the entire variant site is greater than 0.9.
- Ignore broken pairs: When ticked, reads from broken pairs are ignored. Broken pairs may arise for a number of reasons, one being erroneous mapping of the reads. In general, variants based on broken pair reads are likely to be less reliable, so ignoring them may reduce the number of spurious variants called. However, broken pairs may also arise for biological reasons (e.g. due to structural variants) and if they are ignored some true variants may go undetected. Please note that ignored broken pair reads will not be considered for any non-specific match filters.
- Minimum coverage: Only variants in regions covered by at least this many reads are called.
- Minimum count: Only variants that are present in at least this many reads are called.
- Minimum frequency: Only variants that are present at least at the specified frequency (calculated as 'count'/'coverage') are called.
- Specify the parameters for the QC for Targeted Sequencing tool, including a target region file (figure 19.87).
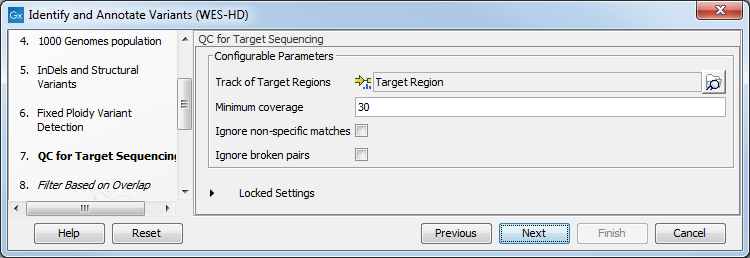
Figure 19.87: Specify the parameters for the QC for Targeted Sequencing tool.The parameters that can be set are:
- Minimum coverage provides the length of each target region that has at least this coverage.
- Ignore non-specific matches: reads that are non-specifically mapped will be ignored.
- Ignore broken pairs: reads that belong to broken pairs will be ignored.
- Specify a targeted region file to remove variants outside of this region.
(figure 19.88)
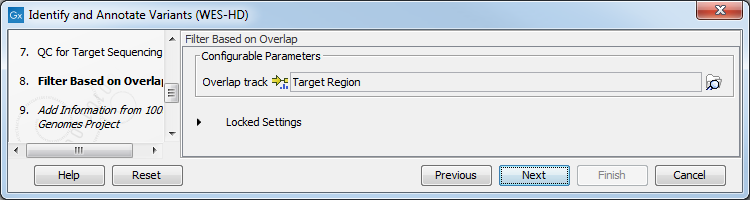
Figure 19.88: Select the targeted region file you used for sequencing. - Specify the 1000 Genomes population that should be used to add information on variants found in the 1000 Genomes project.
- Specify the Hapmap population that should be used to add information on variants found in the Hapmap project.
- In the last wizard step you can check the selected settings by clicking on the button labeled Preview All Parameters.
In the Preview All Parameters wizard you can only check the settings, and if you wish to make changes you have to use the Previous button from the wizard to edit parameters in the relevant windows.
- Choose to Save your results and click on the button labeled Finish.
Output from the Identify and Annotate Variants (WES-HD) workflow
The following outputs are generated:
- A Reads Track
- A Coverage Report Read Mapping
- A Per-region Statistics Track
- A Filtered Variant Track Annotated variants
- An Amino Acid Track Shows the consequences of the variants at the amino acid level in the context of the original amino acid sequence. A variant introducing a stop mutation is illustrated with a red amino acid.
- A Track List