Reads and reference settings
To run the RNA-Seq Analysis for Long Reads tool, go to:
Toolbox | Long Read Support (![]() ) | RNA-Seq Analysis for Long Reads (
) | RNA-Seq Analysis for Long Reads (![]() )
)
Select one or more sequence lists containing long reads.
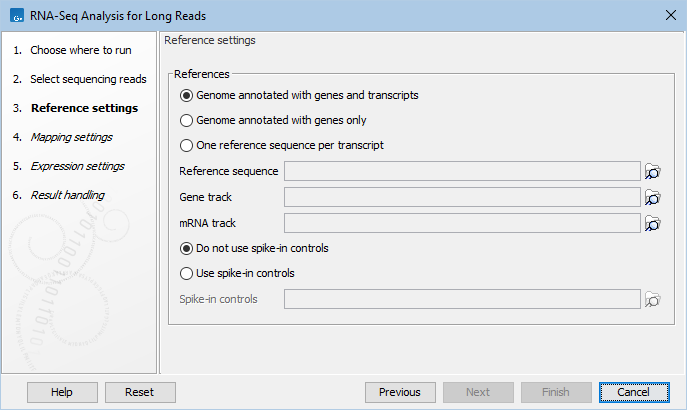
Figure 8.1: RNA-Seq Analysis for Long Reads reference settings.
In the Reference settings dialog (figure 8.1), at the top there are three options concerning how the reference sequences are annotated.
- Genome annotated with genes and transcripts. This option should be used when both gene and mRNA annotations are available. When this option is enabled, the EM will distribute the reads over the transcripts. Gene counts are then obtained by summing over the (EM-distributed) transcript counts. The mRNA annotations are used to define how the transcripts are spliced. This option should be used for Eukaryotes since it is the only option where splicing is taken into account. Note that genes and transcripts are linked by name only (not by position, ID etc).
When this option is selected, both a Gene and an mRNA track should be provided in the boxes below. Annotated reference genomes can be obtained in various ways:
- Directly downloaded as tracks using the Reference Data Manager.
- Imported as tracks from fasta and gff/gtf files.
- Imported from Genbank or EMBL files and converted to tracks.
- Downloaded from Genbank.
When using this option, Expression values, RPKM and TPM are calculated based on the lengths of the transcripts provided by the mRNA track. If a gene's transcript annotation is absent from the mRNA track, all values will be set to zero unless the option "Calculate expression for genes without transcript" is checked in a later dialog.
- Genome annotated with genes only. This option should be used for Prokaryotes where transcripts are not spliced. When this option is selected, a Gene track should be provided in the box below. The data can be obtained in the same ways as described above.
When using this option, Expression values, RPKM and TPM are calculated based on the lengths of the genes provided by the Genes track.
- One reference sequence per transcript. This option is suitable for situations where the reference is a list of sequences. Each sequence in the list will be treated as a "transcript" and expression values are calculated for each sequence. This option is most often used if the reference is a product of a de novo assembly of RNA-Seq data. It is also a suitable option for references where genes are particularly close to each other or clustered in operon structures. When this option is selected, only the reference sequence should be provided, either as a sequence track or a sequence list. Expression values, RPKM and TPM are calculated based on the lengths of sequences from the sequence track or sequence list.
At the bottom of the dialog you can choose between these two options:
- Do not use spike-in controls.
- Use spike-in controls. When selected, you provide a spike-in control file in the field below.
To learn how to import spike-in control files, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Import_RNA_spike_in_controls.html.