SAM and BAM mapping files
The CLC Genomics Workbench supports import and export of files in SAM (Sequence Alignment/Map) and BAM format, which are designed for storing large nucleotide sequence alignments. Read more and see the format specification at http://samtools.sourceforge.net/
The CLC Genomics Workbench includes support for importing SAM and BAM files from Complete Genomics.
Note! If you wish to import the reads in a SAM/BAM file as a sequence list, disregarding any mapping information, please use the Standard import instead.
For a detailed explanation of the SAM and BAM files exported from CLC Genomics Workbench, please see SAM/BAM export format specification.
Input data for importing a mapping from a SAM/BAM file
To import a mapping from a SAM/BAM file containing mapping data into the Workbench, you need to:
- Provide the SAM/BAM file
- Specify the reference sequences that are referred to within that file. The references can either be sequences already imported into the Workbench, or, if appropriately recorded in the SAM/BAM file, can be fetched from URLs specified in the SAM/BAM file.
The mapping is built up within the Workbench using the reference sequence data, the reads and the information from the SAM/BAM file about how the reads are associated with a particular reference.
Data created in the Workbench after importing a SAM/BAM mapping file
- Reads recorded as mapping to a particular reference that is known inside the Workbench are imported as part of the mapping for that reference.
- Reads recorded as not mapping to any reference are imported into a sequence list.
- If they are part of an intact pair, they are imported into a sequence list of paired data.
- If they are single reads or a member of a pair that did not map while its mate did, they are imported into a sequence list containing single reads.
One list is made per read group, with the potential that several such lists could be produced from a single mapping import. The sequence lists are given names of this form for single reads "<read group id> [read group sample] (single) un-mapped reads" and this form for paired reads "<read group id> [read group sample] (paired) un-mapped reads".
- Reads recorded as mapping to a reference sequence that is not known within the Workbench are not imported.
When setting up the import, you are given the option of creating a track-based mapping, or a stand-alone mapping. In the latter case, if there is only one reference sequence, the result will be a single read mapping (![]() ). When there is more than one reference sequence, a multi- mapping object (
). When there is more than one reference sequence, a multi- mapping object (![]() ) is created.
) is created.
Please note that mappings within the CLC Genomics Workbench do not allow for an individual read sequence to map to more than one location. In cases where a SAM/BAM file contains multiple alignment records for a single read, only one such record will be used to build the mapping.
Running the SAM/BAM Mapping Files importer
Click on the Import button on the toolbar or go to:
File | Import (![]() ) | SAM/BAM Mapping Files (
) | SAM/BAM Mapping Files (![]() )
)
This will open a dialog where you select the SAM/BAM file to import as well as the reference sequences to be used (Figure 6.13).
When you select the reference sequence(s) two options exist:
- Select a matching reference sequence that has already been imported into the Workbench. Click on the "Find in folder" icon (
 ) to localize the reference sequence.
) to localize the reference sequence.
- If the SAM/BAM file already contains information about where to find the reference sequence, tick the "Download references" box to automatically download the reference sequence.
The selected reference sequence(s) will be listed under "References in files" with "Name", "Length", and "Status". Whenever the correct reference sequence (with the correct name and sequence length) has been selected the "Status" field will indicate this with an "OK". The length of your reference sequence must match exactly the length of the reference specified in the SAM/BAM file. The name is more flexible as it allows a range of different "synonyms" (with no distinction between capital and lowercase letters). E.g. for chromosome 1 the allowed synonyms would be: 1, chr1, chromosome_1, nc_000001, for chromosome M: m, mt, chrm, chrmt, chromosome_m, chromosome_mt, nc_001807, for chromosome X: x, chrx, chromosome_x, nc_000023, and for chr Y: y, chry, chromosome_y, nc_000024.
If there are inconsistencies in the names or lengths of the reference sequences being chosen and those recorded in the SAM/BAM file, an entry will appear in the "Status" column indicating this. E.g "Length differs" or "Input missing"6.1.
Unmatched reads (reads that are mapped to an unmatched reference e.g. a SAM reference for which there is no CLC reference counterpart) are not imported. The same is the case whenever inconsistencies have occurred with respect to name or length. The log lists all mapping data or unmatched reads that were not imported and marks whether import failed because of unmatched reads being present in the SAM/BAM file or because of inconsistencies in name/length.
Some notes regarding reference sequence naming
Reference sequences in a SAM/BAM file cannot contain spaces. If the name of a reference sequence in the Workbench contains spaces, the Workbench assume that the names of the references in the SAM file will be the same as the names of the References within the Workbench, but with all spaces removed. For exapmple, if your reference sequence in the Workbench was calledmy reference sequence, the Workbench would recognize a reference in the SAM file as the appropriate reference if it was of the same length and had the name myreferencesequence.
Neither the @ character nor the = character are allowed within reference sequence names in SAM files. Any instances of these characters in the name of a reference sequence in the Workbench will be replaced with a _ for the sake of identifying the appropriate reference when importing a SAM or BAM file. For example, if a reference sequence in the Workbench was called my=reference@sequence, the Workbench would recognize a reference in the SAM file as the appropriate reference if it was of the same length and had the name my_reference_sequence.
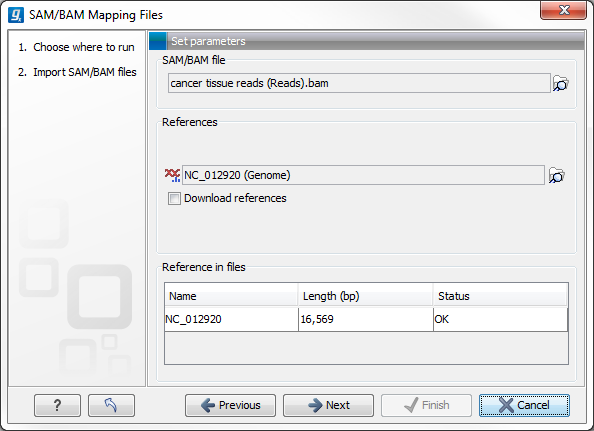
Figure 6.13: Defining SAM/BAM file and reference sequence(s).
Click Next to specify how to handle the results (Figure 6.14). Under Output options the "Save downloaded reference sequence" will be enabled if the "Download references" box was ticked in the previous step (which would be the case when the SAM/BAM file contained information about where to find the reference sequence e.g. if the SAM/BAM file came from an external provider).
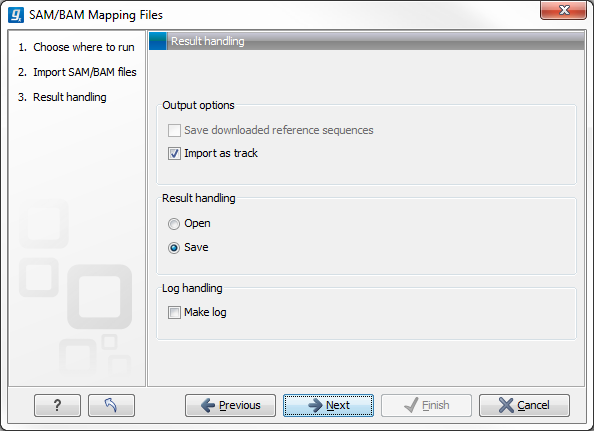
Figure 6.14: Specify the result handling.
Ticking the "Import as track" box results in the generation of a track-based mapping. If the box is not ticked, the file is imported as a standard mapping object.
We recommend choosing Save in order to save the results directly to a folder, as you will probably wish to save the data anyway before proceeding with your analysis. For further information about how to handle the results.
Note that this import operation is very memory-consuming for large data sets, and particularly those with many reads marked as members of broken pairs in the mapping.
Footnotes
- ... missing"6.1
- If you are using a CLC Genomics Server to import files located on the Server (rather than locally), then checks for corresponding reference names and lengths cannot be carried out, so nothing will be reported in this section of the Wizard. This means you will be able to continue to launch the import with correct or incorrect reference sets specified. However, any inconsistencies in these will lead to the import task failing with an error related to this.