How to run the InDels and Structural Variants tool
To start the structural variant detection:
Toolbox | Resequencing (![]() ) | InDels and Structural Variants tool (
) | InDels and Structural Variants tool (![]() )
)
This will open up a dialog. Select the read mapping of interest as shown in figure 26.22 and click on the button labeled Next.
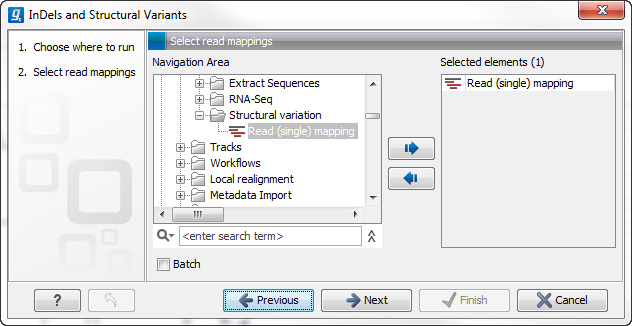
Figure 26.22: Select the read mapping of interest.
The next wizard step (Figure 26.23) is concerned with specifying parameters related to the algorithm used for calling structural variants. The algorithm first identifies positions in the mapping(s) with an excess of reads with left (or right) unaligned ends. Once these positions and the consensus sequences of the unaligned ends are determined, the algorithm maps the determined consensus sequences to the reference sequence around other positions with unaligned ends. If mappings are found that are in accordance with a 'signature' of a structural variant, a structural variant is called. For further details about the algorithm see Section 26.4.3.
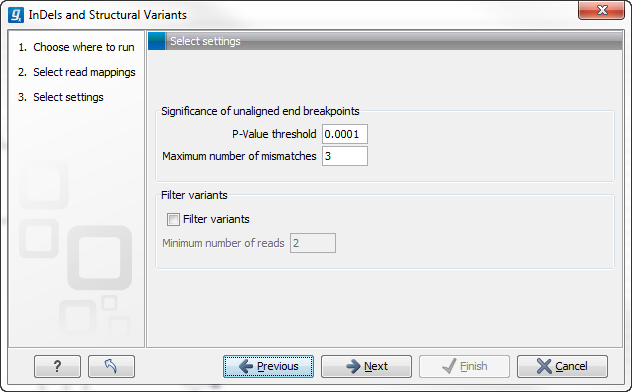
Figure 26.23: Select the relevant settings.
The 'Significance of unaligned end breakpoints' parameters are concerned with when a position with unaligned ends should be considered by the algorithm, and when it should be ignored:
- P-value threshold: Only positions in which the fraction of reads with unaligned ends is sufficiently high will be considered. The 'P-value threshold' determines the cut-off value in a Binomial Distribution for this fraction. The higher the P-value threshold is set, the more unaligned breakpoints will be identified.
- Maximum number of mismatches: The 'Maximum number of mismatches' parameter determines which reads should be considered when inferring unaligned end breakpoints. Poorly map reads tend to have many mis-matches and unaligned ends, and it may be preferable to let the algorithm ignore reads with too many mis-matches in order to avoid false positives and reduce computational time. On the other hand, if the allowed number of mis-matches is set too low, unaligned end breakpoints in proximities of other variants (e.g. SNVs) may be lost. Again, the higher the number of mis-matches allowed, the more unaligned breakpoints will be identified.
The 'Filter variants' parameters are concerned with the amount of evidence for each structural variant required for it to be called:
- Filter variants: When the Filter variants box is checked, only variants that are inferred by breakpoints that together are supported by at least the specified Minimum number of reads will be called.
Specify these settings and click Next. The "Results handling" dialog (Figure 26.24) will be opened. The Indels and Structural variants tool has the following output options:
- Create report When ticked, a report that summarizes information about the inferred breakpoints and variants is created.
- Create breakpoints When ticked, a track containing the detected breakpoints is created.
- Create InDel variants When ticked, a variant track containing the detected InDels that fulfill the requirements for being 'variants' is created. These include the detected insertions for which the allele sequence is inferred, but not those for which it is not, or only partly, known. Also, only deletions of six up to 200 bp are included in the variant track. See Variant tracks for a definition of the requirements for 'variants'. Note that insertions and deletions that are not included in the InDel track, will be present in the 'Structural variants track' (described below).
- Create structural variations When ticked, a track containing the detected structural variants is created.
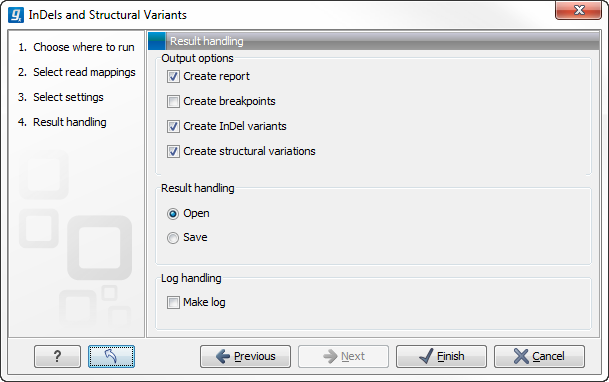
Figure 26.24: Select output formats.
An example of the output from the InDel and Structural Variant tool is shown in Figure 26.25. The output is described in detail in the next section (Section 26.4.2).
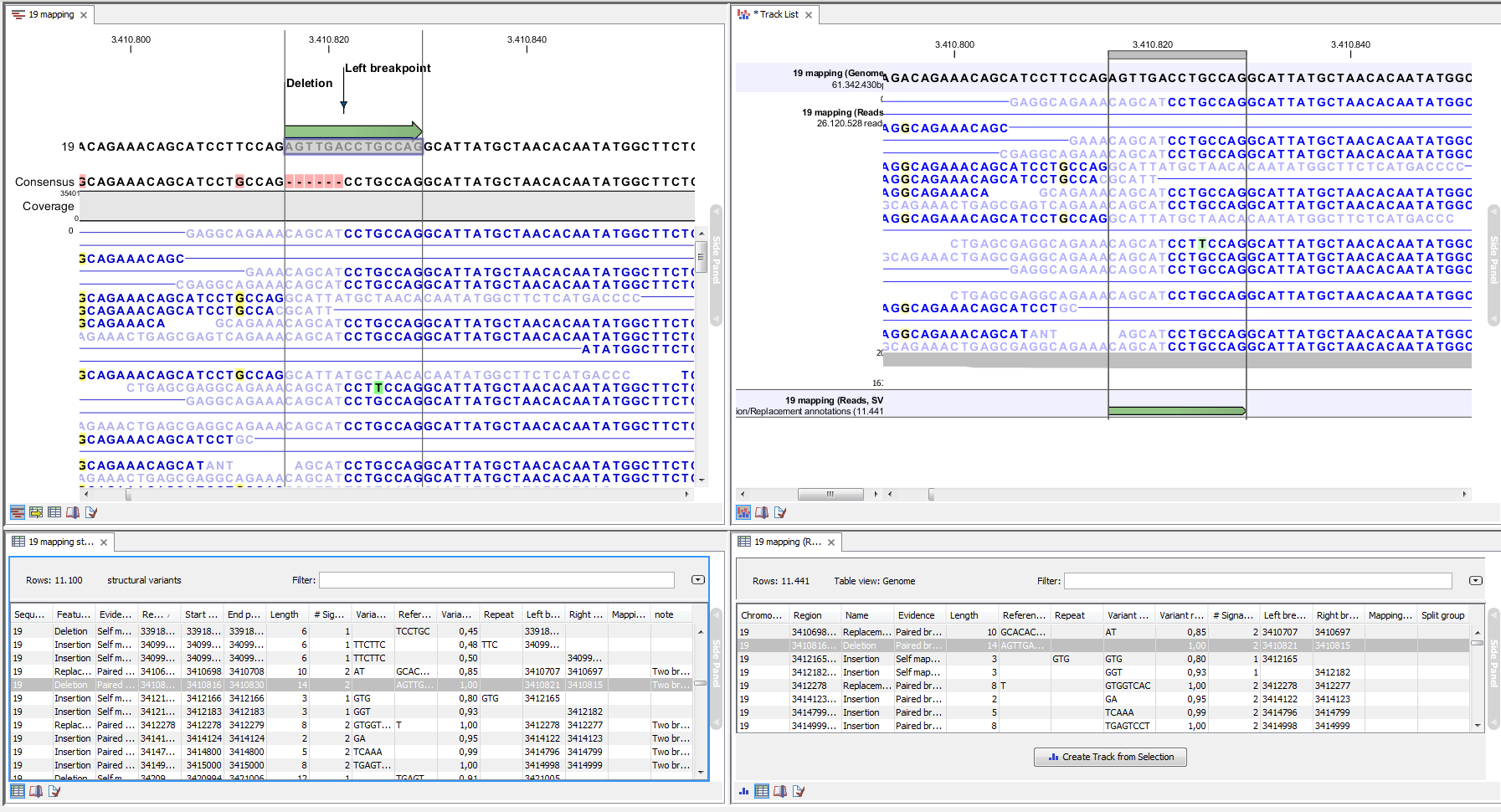
Figure 26.25: Example of the result of an analysis on a standalone read mapping (to the left) and on a reads track (to the right).