RNA-Seq output options
Clicking Next will allow you to specify the output options as shown in figure 27.9.
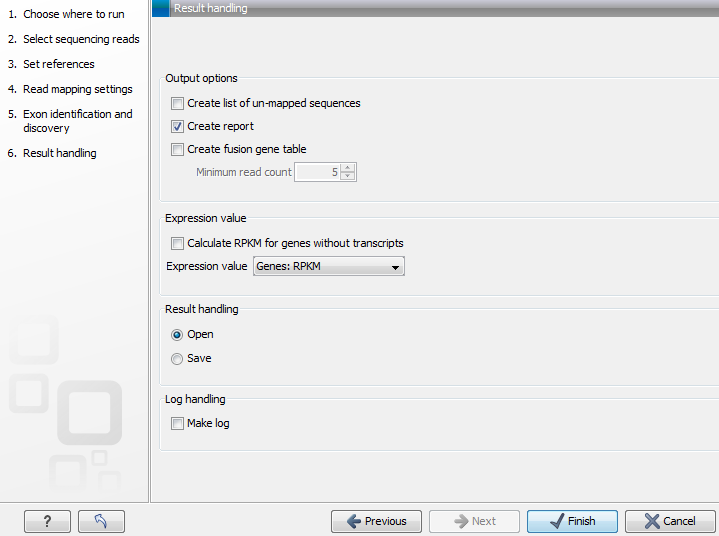
Figure 27.9: Selecting the output of the RNA-Seq analysis.
The Output options are:
- Create list of un-mapped sequences. Creates a list of the un-mapped sequences (marked with a red arrow in figure 27.9). This list can be used to do de novo assembly and perform BLAST searches to see whether you can identify new genes or otherwise further investigate the results.
- Create report. Creates a report of the results. See RNA-Seq report below for a description of the information contained in the report.
- Create fusion gene table. An option that is enabled when using paired data. Creates a table that lists potential fusion genes. This, along with the Minimum read count, is described further below in section Gene fusion reporting.
The standard output for the RNA-Seq analysis is a table showing values for each gene. From the table, mappings can be opened individually by clicking on the button labeled Open mapping found at the bottom of the table or by double clicking on one of the entries in the table (see Interpreting the RNA-Seq analysis result). For eukaryotes, the expression of individual transcripts is also reported.
The expression measure for use in further analysis can be specified under Expression value:
- Calculate RPKM for genes without transcripts. For eukaryotic annotated genomes, specify whether RPKM-like values should be generated for gene features without mRNA annotations. Such features, like small RNAs and tRNAs, have no exons, and thus no exon lengths, which are used in calculating RPKM. When ticked, the "gene length" will be used in place of an "exon length" in the RPKM formula for genes without a corresponding mRNA feature. If this option is not ticked, genes with no mRNA annotations will have given an RPKM value of 0.
- Expression value. The expression measure for use in further analysis can be specified at this point. By default, this is set to Genes RPKM.
The value chosen for measuring expression is used for Interpreting the RNA-Seq analysis result, and for carrying out downstream expression analysis. You can change this to a different value at a later point by opening the result and set the Expression value at the bottom of the table.
Subsections