Defining mapping options for RNA-Seq
When the reference has been defined, click Next and you are presented with the dialog shown in figure 27.5.
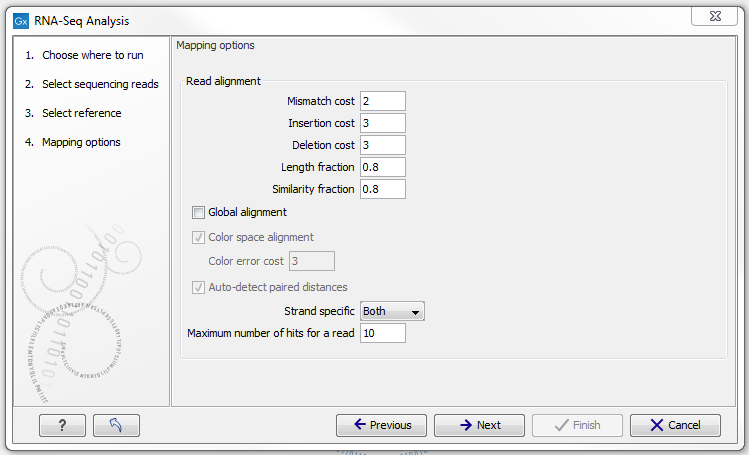
Figure 27.5: Defining mapping parameters for RNA-Seq.
The mapping parameters are identical to those applying to Map Reads to Reference, as the underlying mapping is performed in the same way. For a description of the parameters, please see Mapping parameters.
For the estimation of paired reads distances, RNA-Seq uses the transcript level reference sequence information. This means that introns are not included in the distance measurement. The paired distance measurement will only include transcript sequence, reflecting the true nature of the sequence on which the paired reads were produced.
In addition to the generic mapping parameters, two RNA-Seq specific parameters can be set:
- Maximum number of hits for a read. A read that matches equally well to more distinct places in the references than the 'Maximum number of hits for a read' specified will not be mapped (the notion of distinct places is elaborated below). If a read matches to multiple distinct places, but less than the specified maximum number, it will be randomly assigned to one of these places. The random distribution is done proportionally to the number of unique matches that the genes to which it matches have, normalized by the exon length (to ensure that genes with no unique matches have a chance of having multi-matches assigned to them, 1 will be used instead of 0, for their count of unique matches). This means that if there are 10 reads that match two different genes with equal exon length, the 10 reads will be distributed according to the number of unique matches for these two genes. The gene that has the highest number of unique matches will thus get a greater proportion of the 10 reads.
The definition of a distinct place in the references is complicated because each annotated transcript is extracted and used as reference for the read mapping (if the "Genome annotated with genes and transcripts" is selected in figure 27.4). To exemplify, consider a gene with 10 transcripts and 11 exons, where all transcripts have exon 1, and each of the 10 transcripts have only one of the exons 2 to 11. Exon 1 will be represented 11 times in the references (once for the gene region and once for each of the 10 transcripts). Reads that match to exon 1 will thus match to 11 of the extracted references. However, when the mappings are considered in the coordinates of the main reference genome, it becomes evident that the 11 match places are not distinct but in fact identical. In this case this will just count as one distinct placement of the read, and it will not be discarded for exceeding the maximum number of hits limit. Similarly, when a multi-match read is randomly assigned to one of it's match places, each distinct place is considered only once.
The limit for how many non-specific matches a read is allowed to have, is applied first to the set of gene matches (if any), and then second to the intergenic matches. As an example using the default value of 10, if a read matches equally well 8 places within genes and 50 places in intergenic regions, it is still considered a valid match. It will only be discarded if the number of matches within genes is above the limit, or if there are no gene matches at all and the number of intergenic matches exceeds the limit.
Note that, although a read is mapped distinctly at the gene level, it does not necessarily map uniquely to a particular transcript of the gene. The above example with a gene with 10 transcripts and 11 exons, where all transcripts have exon 1, and each of the 10 transcripts have only one of the exons 2 to 11, is a good an easy to understand example of this: all reads that are mapped to exon 1 are uniquely mapped at the gene level but are non-specific matches at the transcript level. A more complicated example is that you may have a gene with transcript annotations where one transcript has a longer version of an exon than the other. In this case you may have reads that may either be mapped entirely within the long version of the exon, or across the exon-exon boundary of one of the transcripts with the short version of the exon. Such an example is provided by the gene 'Ftl1' in the RNA-seq Mouse Chromosome 7 Tutorial data. The gene and mRNA annotations for that gene are shown in Figure 27.6, along with the reads mapping to the gene.

Figure 27.6: The gene 'Ftl1' from the mouse chromosome 7 tutorial data.When you zoom in on the regions at the end of the second exons and the beginning of the third exons (Figure 27.7) you see that the reference sequence is identical in the start of the part of the second exons that is only present in the long version, and in the start of the third exons (they share the sequence 'CTGCACA'). So a read that is e.g. '...TCATCTTGAGATGGCTTCTGCACA' may be either mapped entirely within the long version of the second exons, or across the exon-exon boundary of the short version of the second exon and the third exon. When it comes to reporting expression levels at the transcript level, reads are randomly assigned among the transcripts to which they map. Note that this introduces some randomness in the numbers of total exon reads for the transcripts (but not for the gene), even when you require that only specific matches are used. Also, as there is the chance that a read may sometimes be assigned to a transcript for which it is an exon-exon read, and sometimes to a transcript for which it is mapped entirely within an exon, even for a run with the maximum number of hits for a read parameter set to 1, there is a random component to the number of exon-exon reads reported, but not to the total number of exon reads.
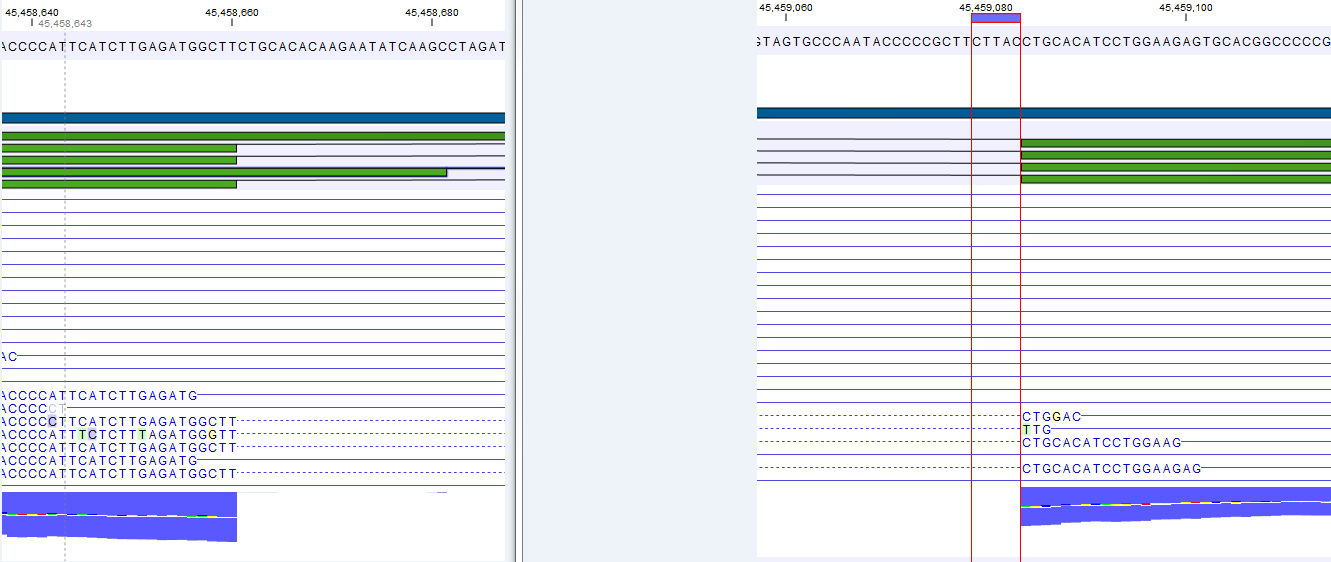
Figure 27.7: The regions at the end of the second exons and the beginning of the third exons of the mRNA transcripts for the gene 'Ftl1'. - Strand-specific alignment. When this option is checked, the user can specify whether the reads should be mapped only in their forward (or reverse) orientation. This will typically be appropriate when a strand specific protocol for read generation has been used. It allows assignment of the reads to the right gene in cases where overlapping genes are located on different strands. Without the strand-specific protocol, this would not be possible (see [Parkhomchuk et al., 2009]). Also, applying the 'strand specific' 'reverse' option in an RNA-seq run could allow the user to assess the degree of antisense transcription. Note that when running RNA seq with the strand specific option turned on you can only make use of pairs in forward-reverse orientation, meaning that mate pairs are not supported.