Identify Somatic Variants from a Single cfDNA Sample using IVA (WES)
To run this workflow, go to:
Toolbox | Ready-to-Use Workflows | Whole Exome Sequencing (![]() ) | Somatic Cancer (
) | Somatic Cancer (![]() ) | Identify Somatic Variants from a Single cfDNA Sample using IVA (WES)
) | Identify Somatic Variants from a Single cfDNA Sample using IVA (WES)
- Double-click on the Identify Somatic Variants from a Single cfDNA Sample using IVA (WES) tool to start the analysis. If you are connected to a server, you will first be asked where you would like to run the analysis.
- Select the sequencing reads (figure 4.9). You can do that by double-clicking on the reads file name or clicking once on the file and then clicking on the arrow pointing to the right side in the middle of the wizard. Click Next.
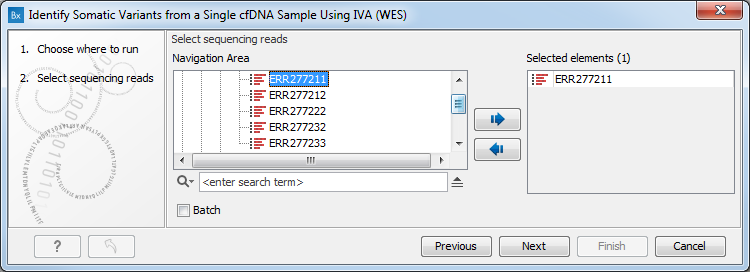
Figure 4.9: Specify the sequencing reads. - Specify the parameters for the Low Frequency Variant Detection tool (figure 4.10).
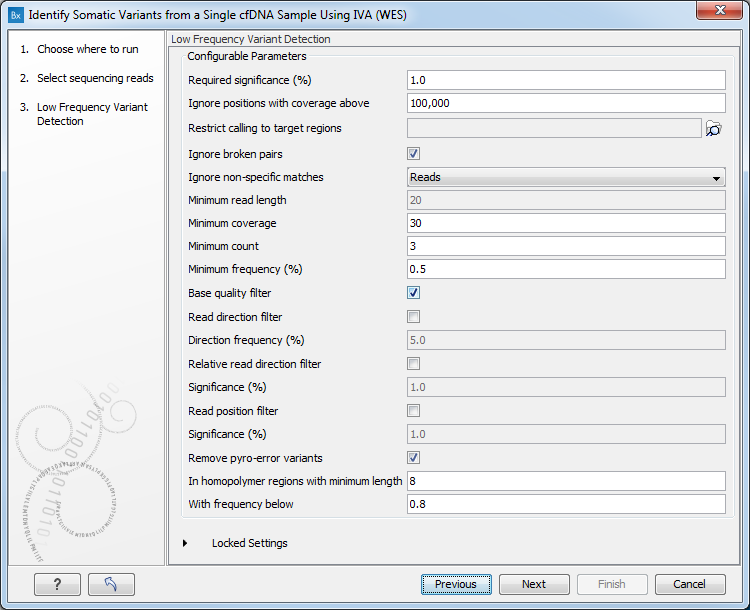
Figure 4.10: Specifying the parameters for the Low Frequency Variant Detection tool.The parameters that can be set are:
- Required significance (%): this parameter determines the cut-off value for the statistical test for the variant not being due to sequencing errors. Only variants that are at least this significant will be called. The lower you set this cut-off, the fewer variants will be called.
- Ignore positions with coverage above: All positions with coverage above this value will be ignored when inspecting the read mapping for variants. The option is highly useful in cases where you have a read mapping which has areas of extremely high coverage as are areas around centromeres in whole genome sequencing applications for example.
- Restrict calling to target regions: Only positions in the regions specified by the target region file will be inspected for variants.
- Ignore broken pairs When ticked, reads from broken pairs are ignored. Broken pairs may arise for a number of reasons, one being erroneous mapping of the reads. In general, variants based on broken pair reads are likely to be less reliable, so ignoring them may reduce the number of spurious variants called. However, broken pairs may also arise for biological reasons (e.g. due to structural variants) and if they are ignored some true variants may go undetected. Please note that ignored broken pair reads will not be considered for any non-specific match filters.
- Ignore non-specific matches You can choose to ignore non-specific matches between reads, regions or to not ignore them at all.
- Minimum read length Only variants in reads longer than this size are called.
- Minimum coverage Only variants in regions covered by at least this many reads are called.
- Minimum count Only variants that are present in at least this many reads are called.
- Minimum frequency Only variants that are present at least at the specified frequency (calculated as 'count'/'coverage') are called.
- Base quality filter: The base quality filter can be used to ignore the reads whose nucleotide at the potential variant position is of dubious quality. This is assessed by considering the quality of the nucleotides in the read in the region around the nucleotide position.
- Read direction filter: The read direction filter removes variants that are almost exclusively present in either forward or reverse reads. For many sequencing protocols such variants are most likely to be the result of amplification induced errors. Note, however, that the filter is NOT suitable for amplicon data, as for this you will not expect coverage of both forward and reverse reads. The filter has a single parameter:
- Direction frequency: Variants that are not supported by at least this frequency of reads from each direction are removed.
- Relative read direction filter: The relative read direction filter attempts to do the same thing as the 'Read direction filter', but does this in a statistical, rather than absolute, sense: it tests whether the distribution among forward and reverse reads of the variant carrying reads is different from that of the total set of reads covering the site. The statistical, rather than absolute, approach makes the filter less stringent. The filter has one parameter:
- Significance: Variants whose read direction distribution is significantly different from the expected with a test at this level, are removed. The lower you set the significance cut-off, the fewer variants will be filtered out.
- Read position filter: The read position filter is a filter that attempts to remove systematic errors in a similar fashion as the 'Read direction filter', but that is also suitable for hybridization-based data. It removes variants that are located differently in the reads carrying it than would be expected given the general location of the reads covering the variant site. This is done by categorizing each sequenced nucleotide (or gap) according to the mapping direction of the read and also where in the read the nucleotide is found; each read is divided in five parts along its length and the part number of the nucleotide is recorded. This gives a total of ten categories for each sequenced nucleotide and a given site will have a distribution between these ten categories for the reads covering the site. If a variant is present in the site, you would expect the variant nucleotides to follow the same distribution. The read position filter carries out a test for whether the read position distribution of the variant carrying reads is different from that of the total set of reads covering the site. The filter has one parameter:
- Significance: Variants whose read position distribution is significantly different from the expected with a test at this level, are removed. The lower you set the significance cut-off, the fewer variants will be filtered out.
- Remove pyro-error variants: This filter can be used to remove insertions and deletions in the reads that are likely to be due to pyro-like errors in homopolymer regions. There are two types of such errors: They may occur either at (1) the immediate ends of homopolymer regions or (2) as an 'overspill' a few nucleotides downstream of a homopolymer region. In case (1) the exact numbers of the same number of nucleotide is uncertain and a sequence like "AAAAAAAA" is sometimes reported as "AAAAAAAAA". In case (2) a sequence like "CGAAAAAGTCG" may sometimes get an 'overspill' insertion of an A between the T and C so that the reported sequence is C "CGAAAAAGTACG". Note that the removal is done in the reads as a very first step, before calling the initial 1 bp variants.
There are two parameters that must be specified for this filter:
- In homopolymer regions with minimum length: Only insertion or deletion variants in homopolymer regions of at least this length will be removed.
- With frequency below: Only insertion or deletion variants whose frequency (ignoring all non-reference and non-homopolymer variant reads) is lower than this threshold will be removed.
- Specify a target region file (figure 4.11). This is a file that depends on the technology you used for sequencing.
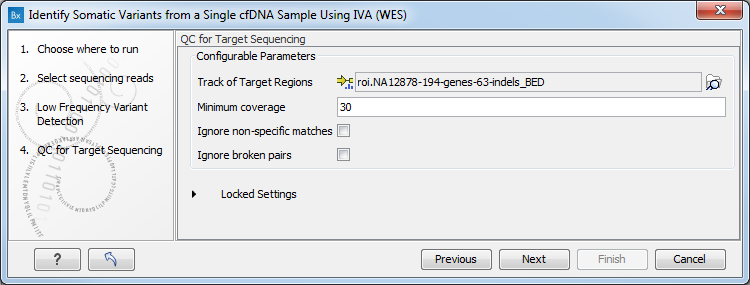
Figure 4.11: Specify a target region file - Specify a variant track for the Identify Known Mutations from Sample Mappings tool (figure 4.12).
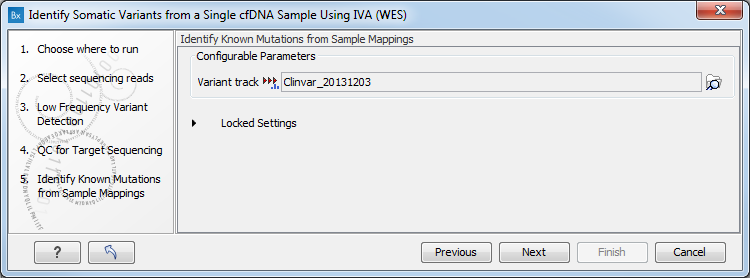
Figure 4.12: Specifying the parameters for the Low Frequency Variant Detection tool.A Clinvar variant track is selected by default, but can be changed to the variant track of your choice if needed.
- In the Ingenuity Variant Analysis dialog, you can specify the Disease name, or leave it to "Any cancer". Enter your login information to Ingenuity Variant Analysis (figure 4.13).
Figure 4.13: Specify login information to Ingenuity Variant Analysis - On the last wizard window, pressing the button Preview All Parameters allows you to preview all parameters. At this step you can only view the parameters, it is not possible to make any changes. Choose to save the results and click on the button labeled Finish.
The following outputs are generated:
- A Reads Track, the read mapping
- A Coverage Report (Target Region Coverage Report)
- A Per-region Statistics Track (Target Region Coverage)
- An Annotated Variant Track, the Identified Known Variants track which contains the known variants provided by the user.
- An Filtered Variant Track for the Somatic Driver Variant, called the Identified Variants
- An Amino Acid Track
- An Imported Variant Track for the Somatic Driver Variant
- A URL file for the Somatic Driver Variant
- A Genome Browser View