Detection filters
The detection filters affect the sensitivity of the Microhaplotype Caller (figure 5.3):
- Required Significance: this parameter determines the cut-off value for the statistical test for the variant not being due to sequencing errors. Only variants that are at least this significant will be called. The lower you set this cut-off, the fewer variants will be called.
- Ignore broken pairs: When enabled, reads from broken pairs are ignored. Broken pairs may arise for a number of reasons, one being erroneous mapping of the reads. In general, variants based on broken pair reads are likely to be less reliable, so ignoring them may reduce the number of spurious variants called. However, broken pairs may also arise for biological reasons (e.g. due to structural variants) and if they are ignored some true variants may go undetected. Please note that ignored broken pair reads will not be considered for any non-specific match filters.
- Non-specific match filter: Non-specific matches are likely to come from repeat region whose exact mapping location is uncertain. In general, variants based on non-specific matches are likely to be less reliable. However, as there are regions in the genome that are entirely perfect repeats, ignoring non-specific matches may have the effect that true variants go undetected in these regions.
There are three options for specifying to which 'extent' the non-specific matches should be ignored:
- 'No': they are not ignored.
- 'Reads': they are ignored.
- 'Region': when this option is chosen no variants are called in regions covered by at least one non-specific match. In this case, the minimum length of reads that are allowed to trigger this effect has to be stated, as really short reads will usually be non-specific even if they do not stem from repeat regions.
- Base quality filter: The base quality filter can be used to ignore the reads whose nucleotide at the potential variant position is of dubious quality. This is assessed by considering the quality of the nucleotides in the region around the nucleotide position. There are three parameters to determine the base quality filter:
- Neighborhood radius: This parameter determines the region size. For example if a neighborhood radius of five is used, a nucleotide will be evaluated based on the nucleotides that are 5 positions upstream and 5 positions downstream of the examined site, for a total of 11 nucleotides. Note that, near the end of the reads, eleven nucleotides will still be considered by offsetting the region relative to the nucleotide in question.
- Minimum central quality: Reads whose central base has a quality below the specified value will be ignored. This parameter does not apply to deletions since there is no 'central base' in these cases.
- Minimum neighborhood quality: Reads for which the minimum quality of the bases is below the specified value will be ignored.
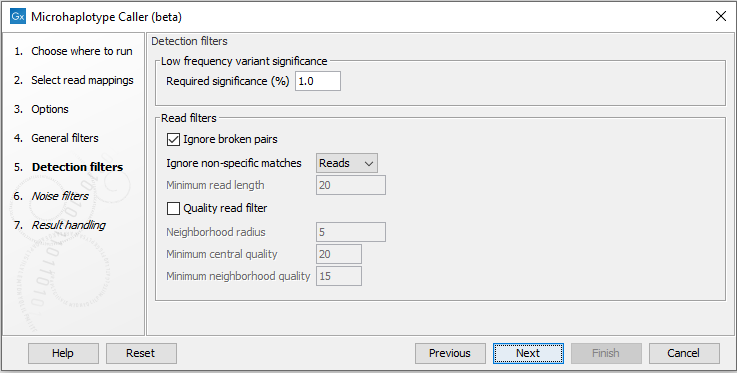
Figure 5.3: The Microhaplotype Caller detection filters.